一、Perl编程优势
1.强大的文本处理能力;
2.快速上手;
3.借鉴其它语言优点;
4.大量的公共模块资源。
二、文件操作&变量的概念
1.第一个perl程序的编写
vim 1.hello.pl
2.完整的程序格式
#!/usr/bin/perl -w use strict
#An example of Perl print "Hello World\n"
/usr/bin/perl 代表的是perl在程序当中的安装路径
-w use strict代表的是提示错误信息
#开头没有!代表的是注释说明作用,不是代码。
3.文件2.readFile.pl的具体操作
(1).test.fa中的文件信息
>t00001 953571
TGTATAGTT
>t00002 781632
TAGCTTATCAGACTGATGTT
>t00003 657683
ACTCGACGCGATATACGTG
(2).具体的函数
#!/usr/bin/perl -w use strict
#Read files
my $file="test.fa";
open(FH,$file) or die $!;
while(<FH>){
print $_;
}
close FH;

3.常见的变量赋值

三、文件处理
1.文件信息处理一
#!/usr/bin/perl -w use strict
#Read files
my $file="test.fa";
open(FH,$file) or die $!;
while(<FH>){
#当读取到某行,匹配行首(^)有">"时, 则跳过这一行不读。
if($_=~/^>/) {next;}
print $_;
}
close FH;
文件程序的运行结果
lhj@lhj-virtual-machine:~$ vim 3.readSeq.pl
lhj@lhj-virtual-machine:~$ perl 3.readSeq.pl
TGTATAGTT
TAGCTTATCAGACTGATGTT
ACTCGACGCGATATACGTG
设计的知识点
正则表达式
正则表达式是在标准格式下,计算机能够匹配的一种模式。
符号: =~//(//内是匹配的内容) =~表示匹配

2.文件信息处理二
(1).统计GC含量
#!/usr/bin/perl -w use strict
#Read files
my $file="test.fa";
open(FH,$file) or die $!;
my $G_num=0;
my $C_num=0;
my $bases=0;
while(<FH>){
if($_=~/^>/) {next;}
#分别计算文件每一行中碱基G,C,以及所有碱基的数目。
if(/C/) {$C_num+=1;}
if(/G/) {$G_num+=1;}
#length($_),计算当前行的长度,这里即每一行的碱基数目。
$bases+=length($_);
}
close FH;
my $GC=($G_num+$C_num)/$bases;
#printf是直接打印双引号中的所有内容
#sprintf则是对于变量进行格式化转变。
$GC=sprintf"%0.2f",$GC; #保留计算结果小数位后两位。
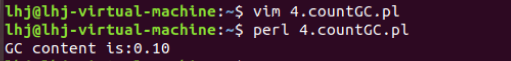
printf "GC content is:$GC\n";
程序运行结果为:
(2).统计每一行基因序列的长度
#!/usr/bin/perl -w use strict
my $file="test.fa";
my %hash;
my $id=0;
open(FH,$file) or die $!;
while(<FH>){
chomp;#代表将每一行后的换行符去掉
if($_=~/^>/) {next;}
#将行的ID和序列长度一一对应存在哈希表%hash
$hash{$id}=length($_);
$id++;
}
close FH;
foreach my $k(sort keys %hash){
#按照ID(sort keys),输出hash。
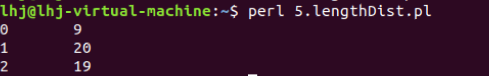
print $k,"\t",$hash{$k},"\n";
}
代码结果:
涉及的知识点:
#!/usr/bin/perl -w
use strict;
#Hash example.
#定义hash时,采用的()切记!
my %hash=(
"Alice"=>100,
"Bill"=>99,
"Cindy"=>79
);
#同数组类似,用foreach循环对hash来读取每个元素。
#keys hash代表的是hash中所有key的集合,每次循环赋值给$k.
foreach my $k(sort keys %hash){
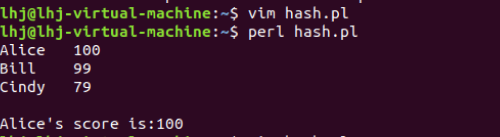
print $k,"\t","$hash{$k}","\n";
}
#读取hash中Alice对应的value值100,表示为$hash{"Alice"}.
print "\nAlice's score is:",$hash{"Alice"},"\n";

程序的运行结果:
(3).代码操作的文件从命令行输入
#!/usr/bin/perl -w use strict
#Read files
my $file=shift;
open(FH,$file) or die $!;
while(<FH>){
chomp;
my @a=split(/\s+/,$_);
if($a[14]<0.5){
print $a[0],"\t",$a[2],"\t",$a[5],"\t",$a[14],"\n";
}
}
close FH;
输入文件为SOAPsnp.out

涉及的知识点:
数组是可以存储多个变量元素(包括字符串、数组),以@为标示符。
数组表示方法:
@bases=("A","C","G","T") @bases=qw(A C G T)
@numbers=(1,2,3,4,5,6,7,8,9) @numbers=(1..10)
数组元素表示:
$base[0]="A"

三、Perl中 调用其它软件
1.比对、基因分析、功能注释
2.使用perl调用其它软件的意义

数据过滤、比对、富集分析、突变检测分析这几个模块,如果要组成一个pipeline一个流程,则需要使用perl将相关的脚本、软件(bwa、soapSNP、GTAK、Samtools)连接起来。
具有如下功能:连接各个模块的功能形成一个pipeline流程,处理这些通用的软件(bwa、soapSNP、GTAK、Samtools),得到下一步的结果。
补充知识点:在linux下运行R
(1).R的代码
#! /usr/lib/R/bin/Rscript --vanilla
c<-scan("5.lengthDist.out")
hist(c)
#options:默认–restore – save --no-readline;–help 查看帮助信息;–version 查看R版本;–slave只打印R脚本的输出,而不显示脚本具体执行情况;–no-timing 去除输出文档结束的运行时间输出。
(2).终端赋予执行权限
$ chmod +x test.r
(3).终端执行脚本
Rscript test.r
如果需要后台挂起可以使用nohup命令
nohup Rscript ./test.r &
在perl下运行R
代码为:
#!/usr/bin/perl -w
use strict;
system("R <test.R --vanilla");
