2022-ACS-Predicting Protein–Ligand Docking Structure with Graph Neural Network

Paper: https://pubs.acs.org/doi/10.1021/acs.jcim.2c00127
Code:https://github.com/j9650/MedusaGraph
用图神经网络预测蛋白质-配体对接结构
本文由美国宾夕法尼亚州立大学Huaipan Jiang课题组2022年发表于ACS文章。
基于对接的药物发现的现有计算软件包存在低准确性和高延迟的问题。通过提高评估蛋白质-配体结合亲和力的能力进行虚拟筛选,但这些方法严重依赖传统的对接软件来对接姿势进行采样,这会导致执行延迟过大。因此,作者提出并评估了一种基于图形神经网络(GNN)的新型框架MedusaGraph,其中包括姿势预测(采样)和姿势选择(评分)模型。
数据
基于PDBbind 2017改进数据集
- 具有少于两个可旋转键的蛋白质-配体复合物以及具有多个配体的蛋白质已被去除
- 除了残基缺失或重复残基的蛋白质。
- 使用 CD-HIT对蛋白质进行聚类(序列同一性截止为 0.9)
最终数据集包含3738个蛋白质-配体复合物
模型

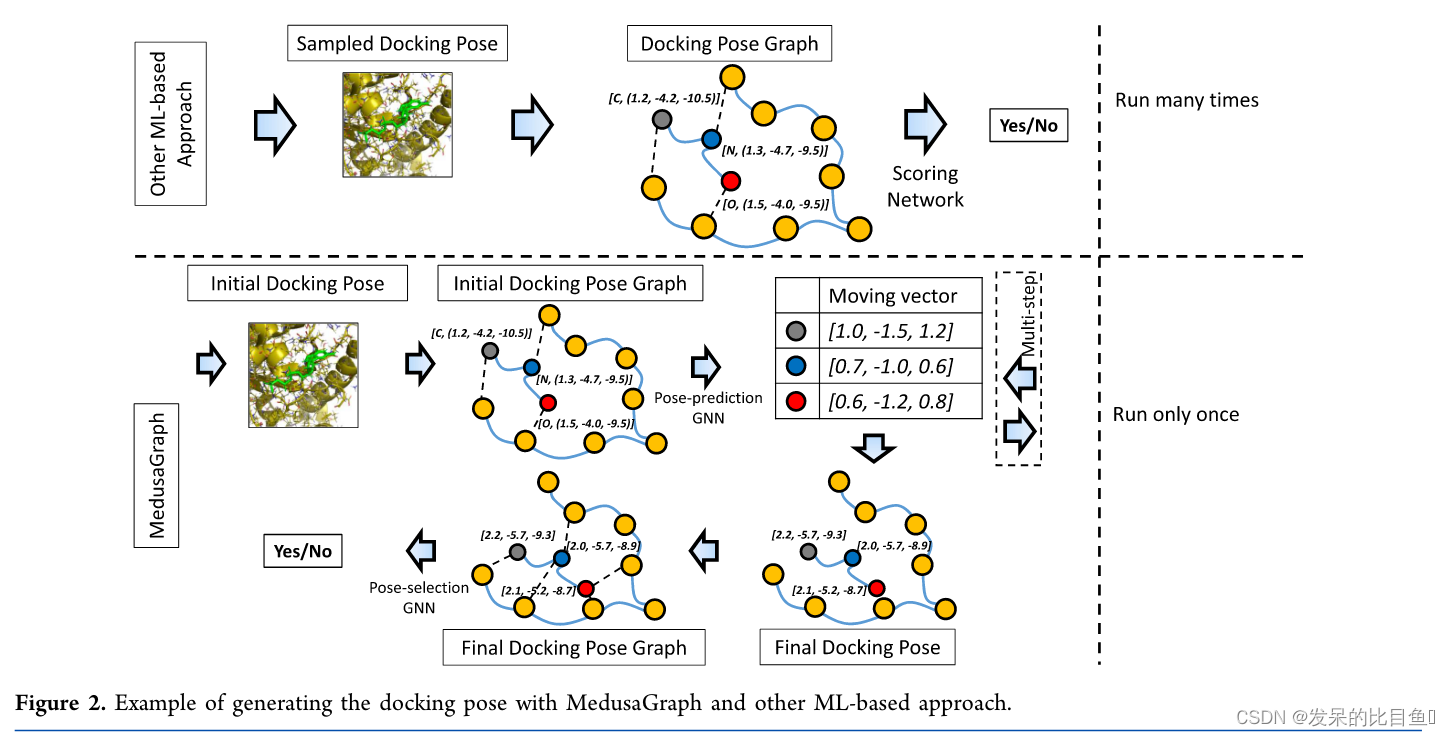
该方法包括2个GNN网络,第一个网络从初始对接姿势预测蛋白质-配体对的最佳对接姿势,第二个网络评估第一个网络的输出姿势并预测姿势是否接近天然。
输入表示
将初始姿势转换为图形表示,其中每个顶点表示复合体中的一个原子,图中的边缘表示节点之间的连接(例如,共价键或附近原子之间的相互作用)。姿势预测模型的输入特征是 N x 21 张量,其中 N 表示复合体中的原子数。每个折点的要素长度为 21。前 18 个元素表示指示原子类型的分类特征。最后三个元素包括初始姿势中原子 ( x , y , z ) (x,y,z) (x,y,z)(x,y,z)的3D坐标。选择 6 Å 阈值作为原子相互作用。图中的边特征包括顶点之间的距离和连接类型(蛋白质-配体、蛋白质-蛋白质或配体-配体)。
模型结构
Graph Neural Network Model.
图中的节点分为刚性节点和柔性节点两部分。姿势预测图神经网络是一种顶点回归模型,它计算柔性节点的运动并输出运动向量 ( x , y , z ) (x,y,z) (x,y,z)(x,y,z),该向量指示沿每个轴的运动。使用 TransformerConv 层来实现这个网络。transformer卷积层采用注意力机制,捕捉每对原子之间的重要性。此外,它还包括边特征(例如,边类型,距离)作为输入特征。TransformerConv 层使用以下等式计算每个节点的输出特征
x i ′ = W 1 x i + ∑ j ∈ N ( i ) α i j ( W 2 x j + W 3 e i j ) x_i^{\prime}=W_1 x_i+\sum_{j \in N(i)} \alpha_{i j}\left(W_2 x_j+W_3 e_{i j}\right) xi′=W1xi+∑j∈N(i)αij(W2xj+W3eij)
其中 x i x_i xi 是节点 i i i 的输入特征向量, x i ′ x_i^′ xi′ 是节点 i i i 的输出特征向量, N ( i ) N(i) N(i) 是节点 i i i 的相邻节点的集合。注意矩阵 α i j α_{ij} αij 是使用
α i j = softmax ( ( W 4 x i ) T ( W 5 x j + W 3 e i j ) d ) \alpha_{i j}=\operatorname{softmax}\left(\frac{\left(W_4 x_i\right)^{\mathrm{T}}\left(W_5 x_j+W_3 e_{i j}\right)}{\sqrt{d}}\right) αij=softmax(d(W4xi)T(W5xj+W3eij))
其中 x i x_i xi表示节点i的输入特征, e i j e_{ij} eij表示边的边特征 ⟨ i , j ⟩ ⟨i,j⟩ ⟨i,j⟩, d d d表示节点特征的隐藏大小;所有 W 都是“可学习”的权重矩阵。使用L1-loss作为损失函数,如下所示
L = ∑ ∣ x c i − x 1 i − x i ∣ + ∣ y c i − y 1 i − y i ∣ + ∣ z c i − z 1 i − z i ∣ L=\sum\left|x_c^i-x_1^i-x^i\right|+\left|y_c^i-y_1^i-y^i\right|+\left|z_c^i-z_1^i-z^i\right| L=∑ xci−x1i−xi + yci−y1i−yi + zci−z1i−zi
其中 ( x i 1 , y i 1 , z i 1 ) (x_i^1, y_i^1, z_i^1) (xi1,yi1,zi1)是第 i i i个原子的初始坐标, ( x i c , y i c , z i c ) (x_i^c, y_i^c, z_i^c) (xic,yic,zic)是X射线晶体结构中第 i i i个原子的坐标, ( x i , y i , z i ) (x_i, y_i, z_i) (xi,yi,zi)是MedusaGraph为第 i i i个原子预测的移动矢量。在训练过程中,只有柔性节点对损失函数有贡献,因为我们只想预测柔性节点的运动。
Multistep Pose Prediction
一种多步姿势预测机制来逐步计算每个原子的最终位置(如图1b所示)。就是将从初始位置到最终位置的路径分为几个步骤,并训练多个模型来预测每个步骤中的原子运动。第 i i i个模型的输出将是第 ( i + 1 ) (i + 1) (i+1)个模型的输入。最后一个模型的输出(对于所有原子)将被视为最终预测的姿势。
Pose Selection
在获得每个复合体的初始对接姿势(图结构)后,将姿势预测GNN应用于初始对接姿势,并获得每个复合体的最终对接姿势。
第二个GNN网络作为姿势选择GNN,将预测这样的姿势是否是一个好的姿势。这个网络基本上是一个图二元分类模型。如图1c所示,我们的模型包括三个TransformerConv层,用于根据其邻居计算每个节点的特征。之后,灵活节点的功能与添加池层一起添加。
结果
MedusaGraph与现有姿势预测方案的比较
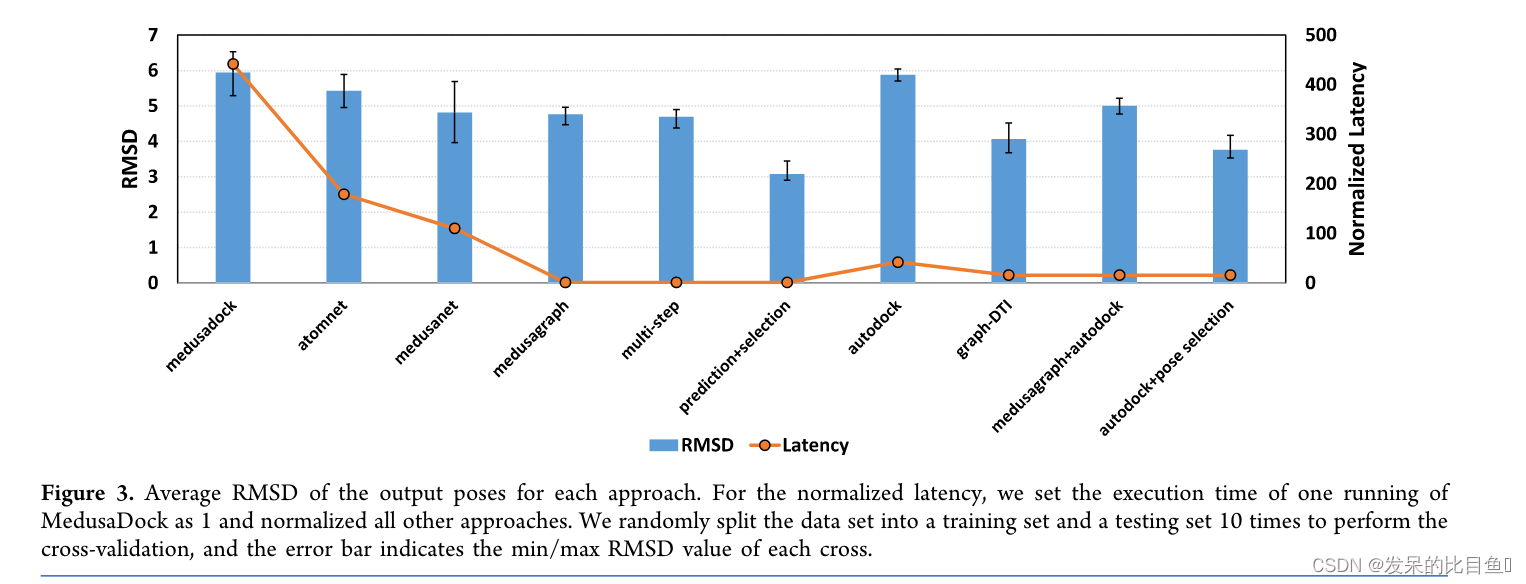
MedusaDock生成的原始姿势中有5.9%的RMSD小于2.5 Å。应用姿势预测模型后,14.4% 的姿势小于 2.5 Å。 使用姿势选择模型,37.6% 的姿势接近原生。
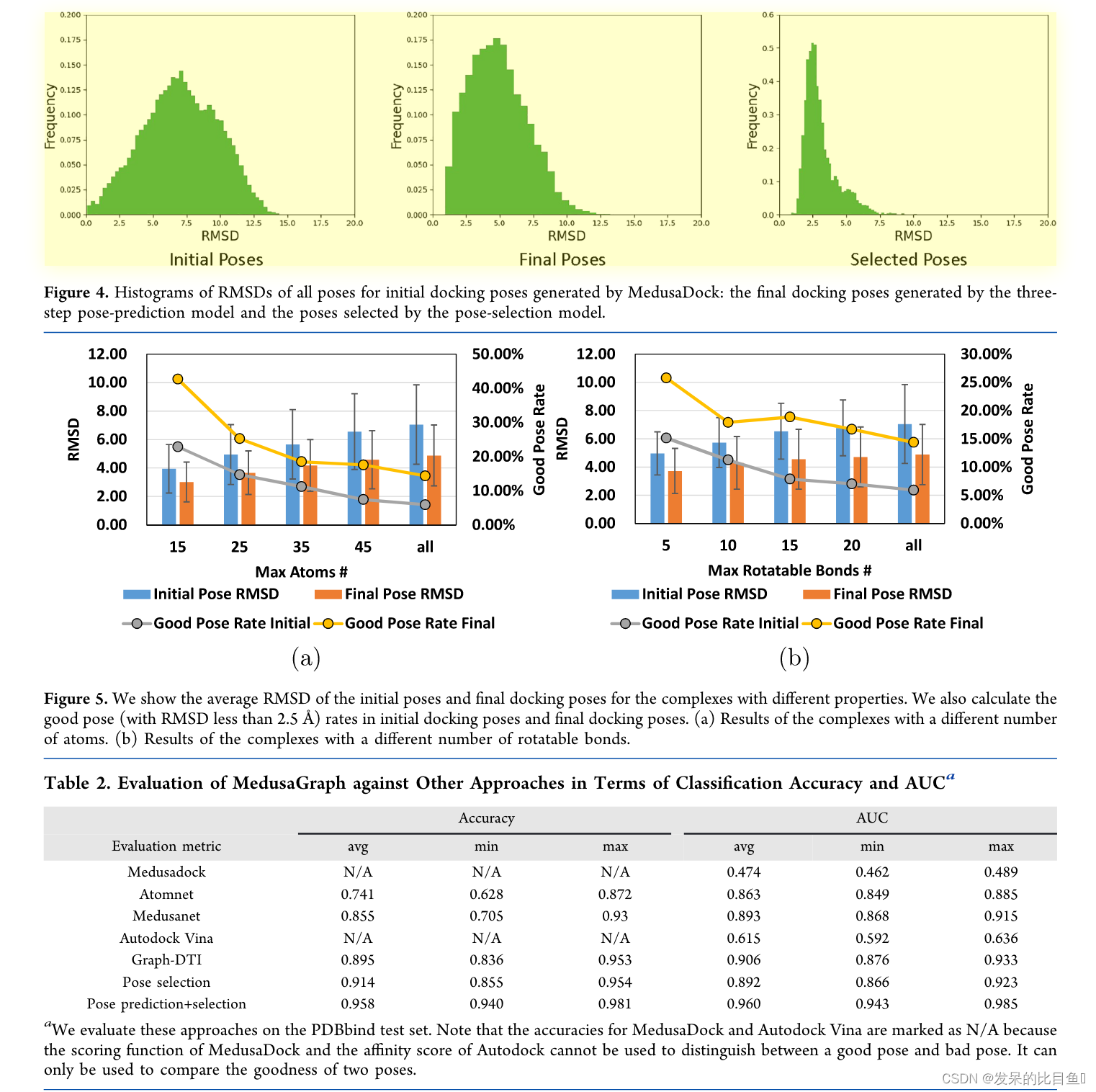
具有不同性质的配体的研究
一些蛋白质-配体复合物比其他复合物更容易找到好的姿势。这主要是因为每个复合体的灵活性可能与其他复合体不同。一般来说,如果配体具有更多的原子,或者配体具有更多的可旋转键,则由此产生的复合物使得找到好的姿势变得更加困难。
姿势选择模型的评估
姿势选择 GNN 模型可以从所有生成的姿势中选择好的姿势,以潜在地改善最终姿势。姿势选择 GNN 模型在姿势预测模型生成的姿势上比 MedusaDock 生成的姿势表现更好,姿势预测 GNN 模型生成的姿势中选择好的姿势比从初始姿势集中选择更容易。

外部数据集评估:CASF
表3中,可以观察到MeusaGraph预测姿势优于其他方法,这表明MeusaGraph可以在药物发现界广泛使用的不同对接功率基准上工作。
[1] Jiang H, Wang J, Cong W, et al. Predicting protein–ligand docking structure with graph neural network[J]. Journal of Chemical Information and Modeling, 2022, 62(12): 2923-2932.