欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://blog.csdn.net/caroline_wendy/article/details/131399818
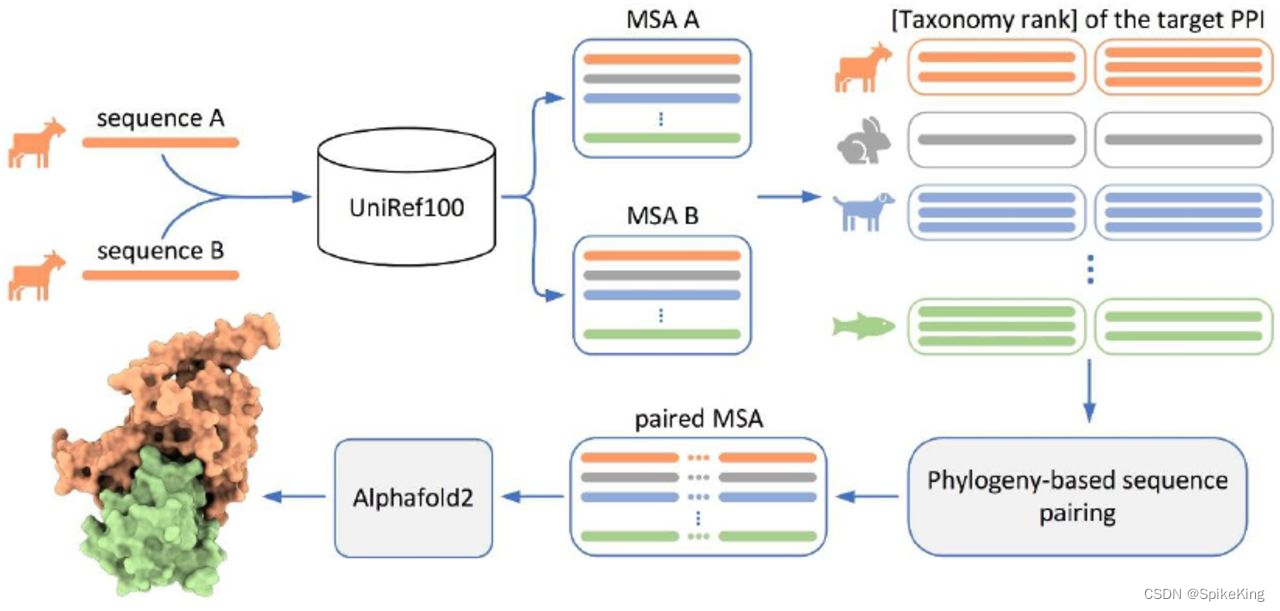
AlphaFold2 Multimer 能够预测多肽链之间相互作用的方法,使用 MSA Pairing 的技术。MSA Pairing 是指通过比较 MSA 来寻找共进化的残基对,这些残基对可能反映了蛋白质之间的接触。AlphaFold2 Multimer 通过计算 MSA Pairing 的得分,来评估不同的多肽链组合的可能性,从而生成最优的三维结构。AlphaFold2 Multimer 提高蛋白质结构预测的准确性和效率,为生物学和医学研究提供了有价值的信息。
官方版本的 AlphaFold2 中 MSA Pairing 的物种信息 (Species) 只处理 uniprot_hits.sto 文件中的信息,其他 MSA 文件不作处理。
1. 解析 sto 文件
stockholm格式是msa的文件格式,解析 sto 文件,提取 MSA 序列和描述,源码如下:
def parse_stockholm(stockholm_string: str):
"""Parses sequences and deletion matrix from stockholm format alignment.
Args:
stockholm_string: The string contents of a stockholm file. The first
sequence in the file should be the query sequence.
Returns:
A tuple of:
* A list of sequences that have been aligned to the query. These
might contain duplicates.
* The deletion matrix for the alignment as a list of lists. The element
at `deletion_matrix[i][j]` is the number of residues deleted from
the aligned sequence i at residue position j.
* The names of the targets matched, including the jackhmmer subsequence
suffix.
"""
name_to_sequence = collections.OrderedDict()
for line in stockholm_string.splitlines():
line = line.strip()
if not line or line.startswith(('#', '//')):
continue
name, sequence = line.split()
if name not in name_to_sequence:
name_to_sequence[name] = ''
name_to_sequence[name] += sequence
msa = []
deletion_matrix = []
query = ''
keep_columns = []
for seq_index, sequence in enumerate(name_to_sequence.values()):
if seq_index == 0:
# Gather the columns with gaps from the query
query = sequence
keep_columns = [i for i, res in enumerate(query) if res != '-']
# Remove the columns with gaps in the query from all sequences.
aligned_sequence = ''.join([sequence[c] for c in keep_columns])
msa.append(aligned_sequence)
# Count the number of deletions w.r.t. query.
deletion_vec = []
deletion_count = 0
for seq_res, query_res in zip(sequence, query):
if seq_res != '-' or query_res != '-':
if query_res == '-':
deletion_count += 1
else:
deletion_vec.append(deletion_count)
deletion_count = 0
deletion_matrix.append(deletion_vec)
return msa, list(name_to_sequence.keys())
stockholm格式文件的描述信息,主要关注于 GS 至 DE 之间的部分,类似 a3m 中的 > 描述:
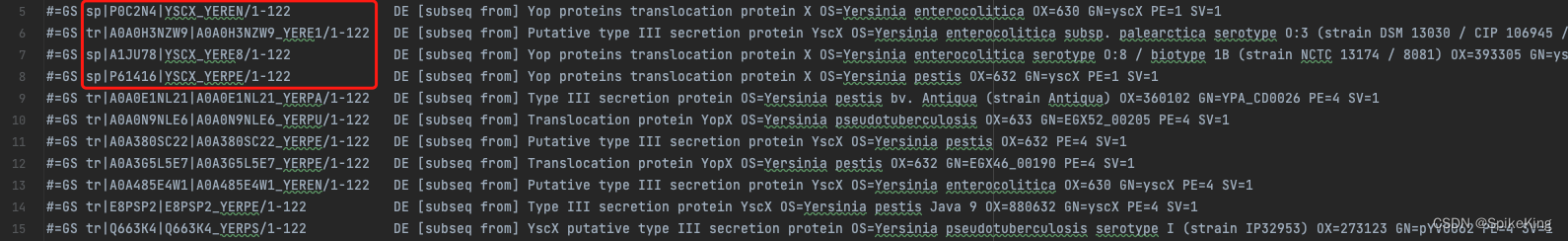
解析出的信息如下:
[Info] sample 173 desc sp|P0C2N4|YSCX_YEREN/1-122, seq MSRIITAPHIGIEKLSAISLEELSCGLPDRYALPPDGHPVEPHLERLYPTAQSKRSLWDFASPGYTFHGLHRAQDYRRELDTLQSLLTTSQSSELQAAAALLKCQQDDDRLLQIILNLLHKV
[Info] species: YEREN
[Info] per_seq_similarity: 1.0
[Info] sample 173 desc tr|A0A0H3NZW9|A0A0H3NZW9_YERE1/1-122, seq MSRIITAPHIGIEKLSAISLEELSCGLPDRYALPPDGHPVEPHLERLYPTAQSKRSLWDFASPGYTFHGLHRAQDYRRELDTLQSLLTTSQSSELQAAAALLKCQQDDDRLLQIILNLLHKV
[Info] species: YERE1
[Info] per_seq_similarity: 1.0
[Info] sample 173 desc sp|A1JU78|YSCX_YERE8/1-122, seq MSRIITAPHIGIEKLSAISLEELSCGLPERYALPPDGHPVEPHLERLYPTAQSKRSLWDFASPGYTFHGLHRAQDYRRELDTLQSLLTTSQSSELQAAAALLKCQQDDDRLLQIILNLLHKV
[Info] species: YERE8
[Info] per_seq_similarity: 0.9918032786885246
[Info] sample 173 desc sp|P61416|YSCX_YERPE/1-122, seq MSRIITAPHIGIEKLSAISLEELSCGLPERYALPPDGHPVEPHLERLYPTAQSKRSLWDFASPGYTFHGLHRAQDYRRELDTLQSLLTTSQSSELQAAAALLKCQQDDDRLLQIILNLLHKV
[Info] species: YERPE
[Info] per_seq_similarity: 0.9918032786885246
2. 提取物种信息
使用序列描述提取物种信息,即|所分割的第3部分中的后半段,源码如下:
- 例如描述是
sp|P0C2N4|YSCX_YEREN/1-122,物种是YEREN
# Sequences coming from UniProtKB database come in the
# `db|UniqueIdentifier|EntryName` format, e.g. `tr|A0A146SKV9|A0A146SKV9_FUNHE`
# or `sp|P0C2L1|A3X1_LOXLA` (for TREMBL/Swiss-Prot respectively).
_UNIPROT_PATTERN = re.compile(
r"""
^
# UniProtKB/TrEMBL or UniProtKB/Swiss-Prot
(?:tr|sp)
\|
# A primary accession number of the UniProtKB entry.
(?P<AccessionIdentifier>[A-Za-z0-9]{6,10})
# Occasionally there is a _0 or _1 isoform suffix, which we ignore.
(?:_\d)?
\|
# TREMBL repeats the accession ID here. Swiss-Prot has a mnemonic
# protein ID code.
(?:[A-Za-z0-9]+)
_
# A mnemonic species identification code.
(?P<SpeciesIdentifier>([A-Za-z0-9]){1,5})
# Small BFD uses a final value after an underscore, which we ignore.
(?:_\d+)?
$
""",
re.VERBOSE)
def _parse_sequence_identifier(msa_sequence_identifier: str):
"""Gets species from an msa sequence identifier.
The sequence identifier has the format specified by
_UNIPROT_TREMBL_ENTRY_NAME_PATTERN or _UNIPROT_SWISSPROT_ENTRY_NAME_PATTERN.
An example of a sequence identifier: `tr|A0A146SKV9|A0A146SKV9_FUNHE`
Args:
msa_sequence_identifier: a sequence identifier.
Returns:
An `Identifiers` instance with species_id. These
can be empty in the case where no identifier was found.
"""
matches = re.search(_UNIPROT_PATTERN, msa_sequence_identifier.strip())
if matches:
return matches.group('SpeciesIdentifier')
return ""
def _extract_sequence_identifier(description: str):
"""Extracts sequence identifier from description. Returns None if no match."""
split_description = description.split()
if split_description:
return split_description[0].partition('/')[0]
else:
return None
def get_identifiers(description: str):
"""Computes extra MSA features from the description."""
sequence_identifier = _extract_sequence_identifier(description)
if sequence_identifier is None:
return ""
else:
return _parse_sequence_identifier(sequence_identifier)
3. 计算序列相似度
按照氨基酸一致性百分比计算相似度,完全相同是1.0,源码如下:
def msa_seq_similarity(query_seq, msa_seq):
"""
MSA 序列相似度
"""
query_seq = np.array(list(query_seq))
msa_seq = np.array(list(msa_seq))
seq_similarity = np.sum(query_seq == msa_seq) / float(len(query_seq))
return seq_similarity
测试脚本,如下:
class MultimerSpeciesParser(object):
def __init__(self):
pass
def process(self, a_path, b_path):
assert os.path.isfile(a_path)
assert os.path.isfile(b_path)
print(f"[Info] A 链路径: {
a_path}")
print(f"[Info] B 链路径: {
b_path}")
with open(a_path) as f:
sto_string = f.read()
a_msa, a_desc = parse_stockholm(sto_string)
assert len(a_msa) == len(a_desc)
for i in range(1, 5):
print(f"[Info] sample {
len(a_msa)} desc {
a_desc[i]}, seq {
a_msa[i]}")
sid = get_identifiers(a_desc[i])
print(f"[Info] species: {
sid}")
per_seq_similarity = msa_seq_similarity(a_msa[0], a_msa[i])
print(f"[Info] per_seq_similarity: {
per_seq_similarity}")