Spateo工具使用指南(一)
近期发现Spateo这个神奇的工具能做到细胞分割,里面也有很多有趣的算法
安装
值得注意的是win系统目前安装不了!!!方法仅适用于linux系统!!!
我们需要至少3.7及以上版本的python。
#这里我们使用3.9版本的python
conda create -n spa python=3.9
有三种方式安装该包,(pip/pip git/离线)但是实测各种版本冲突之下只有pip有效,下面是代码:
#方法一
pip install spateo-release
方法二(有网络条件的可以自己尝试一下)
pip install git+https://github.com/aristoteleo/spateo-release
官方的 Known Issues and Fixes
如果有冲突可以运行
pip install h5py==3.7.0
pip install anndata==0.8.0
值得注意的是win系统目前安装不了!!!以上方法仅适用于linux系统!!!
步入正题,使用Spateo进行细胞染色质分割
在本教程中,我们假设我们已经为同一组织切片配对了 RNA 和细胞核染色图像,并且这两个坐标系(粗略地)配准了。我们将使用细胞核染色作为细胞核的真实位置,并使用此信息获得细胞分割。我们将在以下步骤中这样做。
1.作为预处理步骤,我们将改进染色图像和 RNA 坐标之间的对齐。
2.使用基于分水岭的方法识别和标记单个细胞核。
3.使用称为StarDist的基于深度学习的方法执行相同的操作。
4.[可选] 通过复制分水岭方法中存在但不与 StarDist 方法中的任何标签重叠的标签,使用分水岭标签增强 StarDist 标签。
5.[可选] 将细胞核标签扩展到细胞质。
导入安装好的包
作者用的应该是苹果电脑
import spateo as st
import matplotlib.pyplot as plt
st.config.n_threads = 8
%config InlineBackend.print_figure_kwargs = {
'facecolor' : "w"}
%config InlineBackend.figure_format = 'retina'
加载数据
我们将使用Chen 等人于 2021 年截断的小鼠冠状切面数据集。这是一个提供 ssDNA(细胞核)染色的 Stereo-seq 数据集。
!wget "https://drive.google.com/uc?export=download&id=1nONOaUy7utvtXQ3ZPx7R3TePq2Oo4JFM" -nc -O SS200000135IL-D1.ssDNA.tif
!wget "https://drive.google.com/uc?export=download&id=18sM-5LmxOgt-3kq4ljtq_EdWHjihvPUx" -nc -O SS200000135TL_D1_all_bin1.txt.gz
将下载的 UMI 计数和核染色图像加载到 AnnData 对象中。出于细胞分割的目的,我们将使用聚合计数矩阵,其中AnnData 的obs和var对应于空间 X 和 Y 坐标,矩阵的每个元素包含为每个 X 和 Y 捕获的 UMI 总数协调。
adata = st.io.read_bgi_agg(
'SS200000135TL_D1_all_bin1.txt.gz', 'SS200000135IL-D1.ssDNA.tif',
)
adata
|-----> 构建计数矩阵。
|-----> __type 到 AnnData 对象中的 uns。
|-----> pp 到 AnnData 对象中的 uns。
|-----> 空间到 AnnData 对象中的 uns。
具有 n_obs × n_vars = 2000 × 2000 的 AnnData 对象
uns: ‘__type’, ‘pp’, ‘空间’
图层:“染色”、“拼接”、“未拼接”
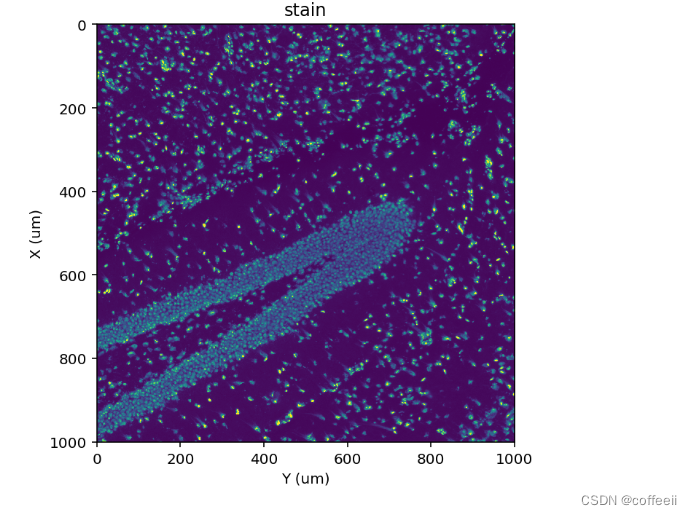
st.pl.imshow(adata, 'stain')
|-----> AnnData 对象中的 染色层
优化对齐
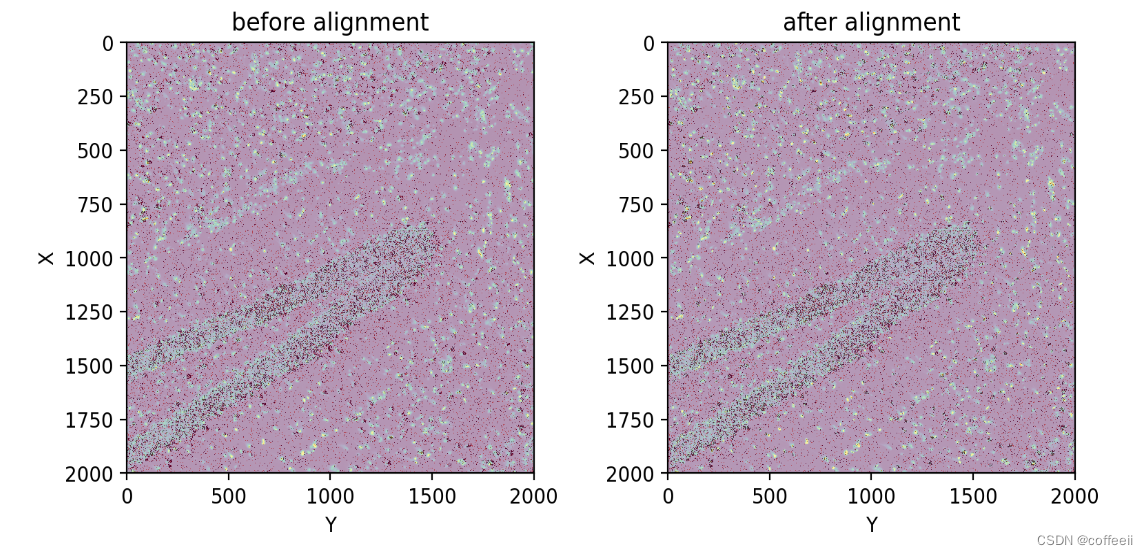
染色图像应该已经与 RNA 坐标大致对齐,但可能会有轻微的错位。大的错位会导致不正确的 UMI 聚合(因此不正确的单元格!)。因此,我们认为改进直接从空间转录组学分析提供的比对是一种很好的做法。
Spateo 提供了两种比对策略,但在这里我们将使用更简单的rigid比对,因为测试表明这对这个样本表现良好。对于其他样本,non-rigid对齐方法可能表现更好。
before = adata.layers['stain'].copy()
st.cs.refine_alignment(adata, mode='rigid', transform_layers=['stain'])
|-----> AnnData 对象中的 染色层
|-----> AnnData 对象中的未拼接层
|-----> 在刚性模式下细化对齐。
|-----> 变换图层 [‘stain’]
|-----> AnnData 对象中的 染色层
|-----> 对 AnnData 对象中的层进行染色。
fig, axes = plt.subplots(ncols=2, figsize=(8, 4), tight_layout=True)
axes[0].imshow(before)
st.pl.imshow(adata, 'unspliced', ax=axes[0], alpha=0.6, cmap='Reds', vmax=2, use_scale=False, save_show_or_return='return')
axes[0].set_title('before alignment')
st.pl.imshow(adata, 'stain', ax=axes[1], use_scale=False, save_show_or_return='return')
st.pl.imshow(adata, 'unspliced', ax=axes[1], alpha=0.6, cmap='Reds', vmax=2, use_scale=False, save_show_or_return='return')
axes[1].set_title('after alignment')
|-----> AnnData 对象中的未拼接层
|-----> AnnData 对象中的 染色层
|-----> AnnData 对象中的未拼接层
Text(0.5, 1.0, ‘对齐后’)
基于分水岭的方法
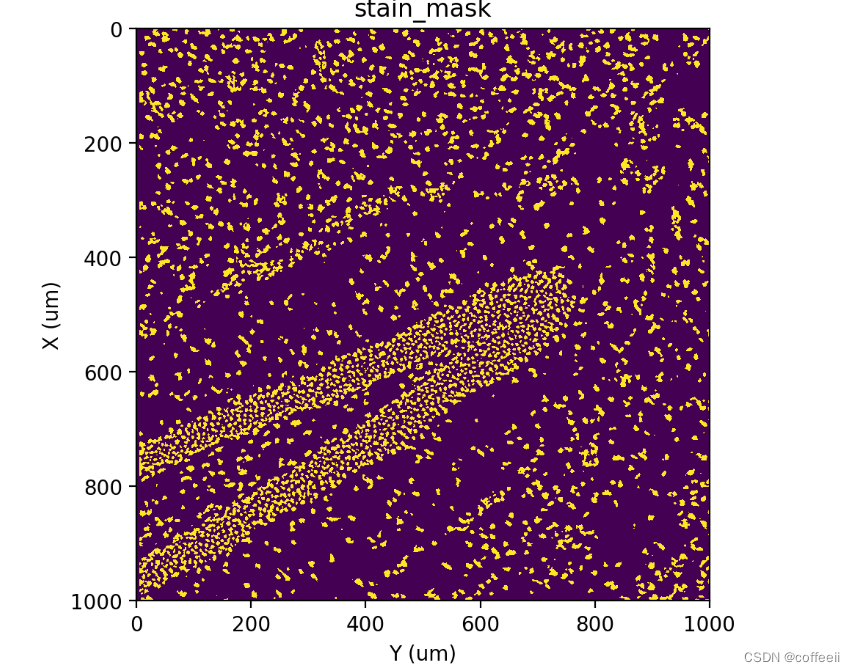
Spateo 包括一种基于分水岭的自定义方法,用于从染色图像中分割和标记细胞核。在高层次上,它使用全局和局部阈值组合首先获得细胞核掩码(回想一下这称为分割),然后使用 Watershed 分配标签(回想一下这称为标记)。
分割
st.cs.mask_nuclei_from_stain(adata)
st.pl.imshow(adata, 'stain_mask')
|-----> AnnData 对象中的 染色层
|-----> 从染色图像构建核掩模。
|-----> stain_mask 到 AnnData 对象中的图层。
|-----> AnnData 对象中的 stain_mask 层
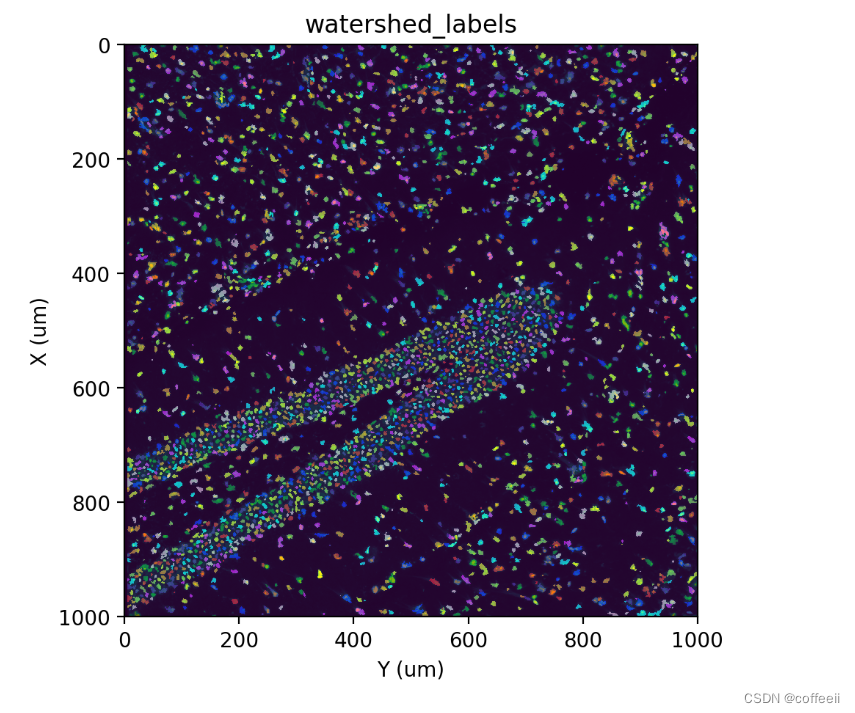
标签
st.cs.find_peaks_from_mask(adata, 'stain', 7)
st.cs.watershed(adata, 'stain', 5, out_layer='watershed_labels')
fig, ax = st.pl.imshow(adata, 'stain', save_show_or_return='return')
st.pl.imshow(adata, 'watershed_labels', labels=True, alpha=0.5, ax=ax)
|-----> AnnData 对象中的 stain_mask 层
|-----> 查找最小距离为 7 的峰。
|-----> 到 AnnData 对象中图层的 stain_distances。
|-----> stain_markers 到 AnnData 对象中的图层。
|-----> AnnData 对象中的 染色层
|-----> AnnData 对象中的 stain_mask 层
|-----> AnnData 对象中的 stain_markers 层
|-----> 流动分水岭。
|-----> watershed_labels 到 AnnData 对象中的图层。
|-----> AnnData 对象中的 染色层
|-----> AnnData 对象中的 watershed_labels 层
截止目前就搞定了!但我们还需要更多的了解其数据结构!需要注意的是若有不同的读入结构可能没有spliced和unspliced层。
更多生信知识欢迎交流v:coffeeiix(也可接单细胞转录组分析培训)