版权声明:本文为博主原创文章,未经博主允许不得转载。 https://blog.csdn.net/u012325865/article/details/82153557
基于Python3和RDKit的化合物结构相似性搜索
化合物相似性在化学信息学和药物发现中具有悠久的历史,许多计算方法采用相似度测定来鉴定研究的新化合物。
本实例通过计算分子的Morgan指纹进行相似性比对。
代码实例:
#导入依赖包
#!/usr/bin/env python3
from rdkit.Chem import AllChem as ch
from rdkit.Chem import Draw as d
from rdkit import DataStructs#载入分子库
suppl = ch.SDMolSupplier('drugbank.sdf')
mols = [x for x in suppl if x is not None]
len(mols) #计算分子库分子数目#读入查询分子,计算指纹
nicotine = ch.MolFromSmiles('O=C(C)Oc1ccccc1C(=O)O')
nicotine_fingerprint = ch.GetMorganFingerprint(nicotine, 2)#计算分子库每个分子指纹
mols_fps = [(m, ch.GetMorganFingerprint(m, 2)) for m in mols]#计算相似度并排序,输出最相似的的前20个分子
mols_nicotinesim = [(m, DataStructs.TanimotoSimilarity(fp, nicotine_fingerprint))
for m, fp in mols_fps]
sorted_mols_nicotinesim = sorted(mols_nicotinesim, key=lambda x: x[1], reverse=True)
result = sorted_mols_nicotinesim[:20]#输出最相似的前20个结构
map(ch.Compute2DCoords, (m for m, sim in result))
img = d.MolsToGridImage([m for m, sim in result], kekulize=False, subImgSize=(400,400),
legends=[mol.GetProp("GENERIC_NAME") + ': ' + str(sim)
for mol, sim in result])
img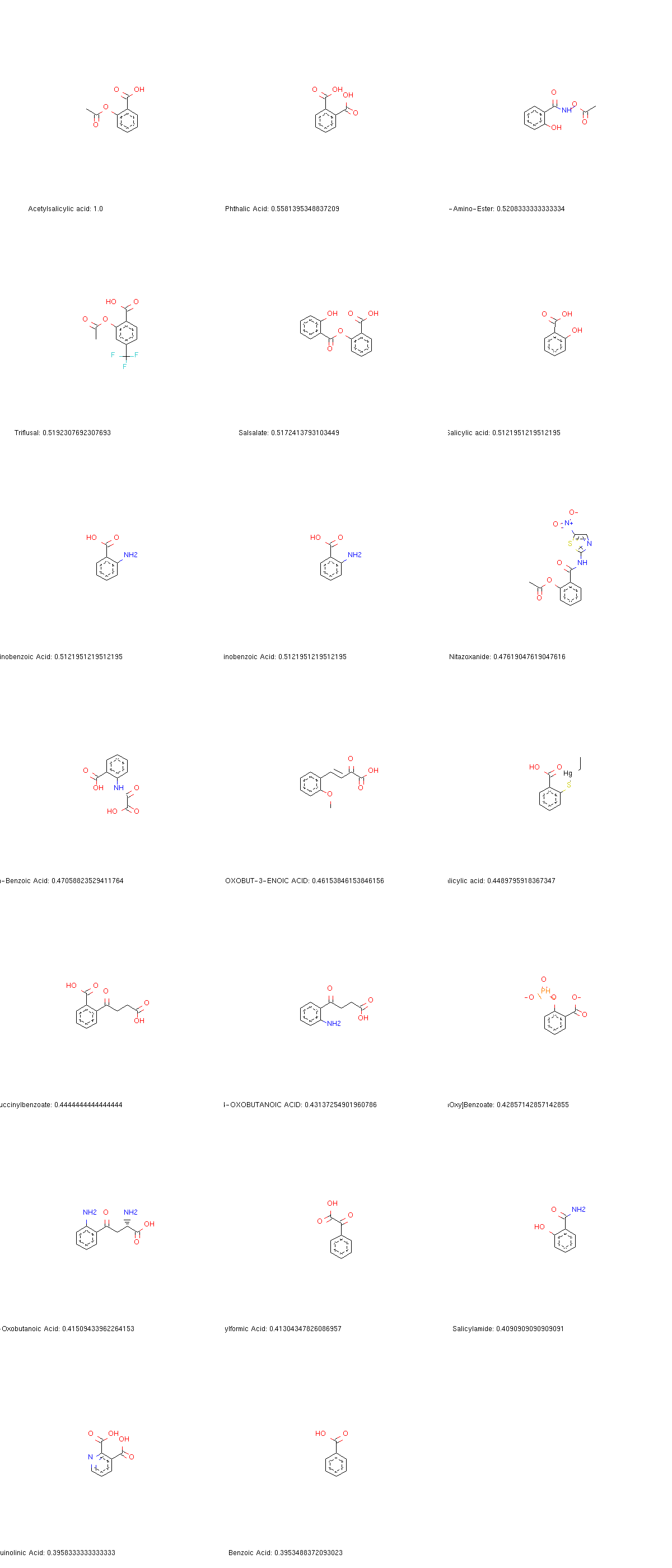
注:代码中用到的文件需要可以留言博主