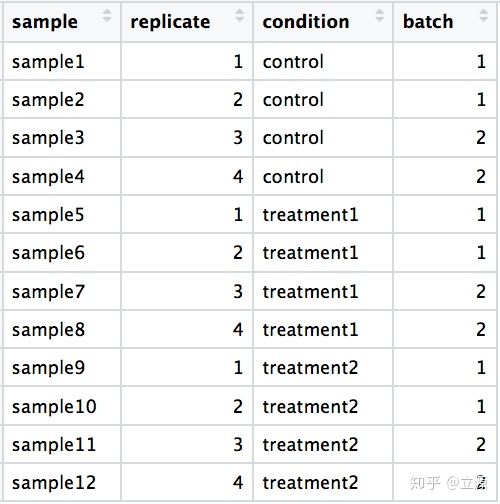
实验设计时应该考虑的因素
理解如何提取RNA和RNA-Seq建库的实验步骤对于设计一个RNA-Seq实验是非常有帮助的,但是有一些可以严重影响差异表达分析质量的特殊因素应该被考虑。
这些重要的考虑因素包括:
- 重复(replicates)的数目和类型
- 避免混淆(confounding)
- 处理批次效应(batch effects)
我们将详细浏览每一个考虑因素,并讨论最佳实践和优化设计
重复(Replicates)
实验重复包括技术性重复(technical replicates)和生物学重复(biological replicates)
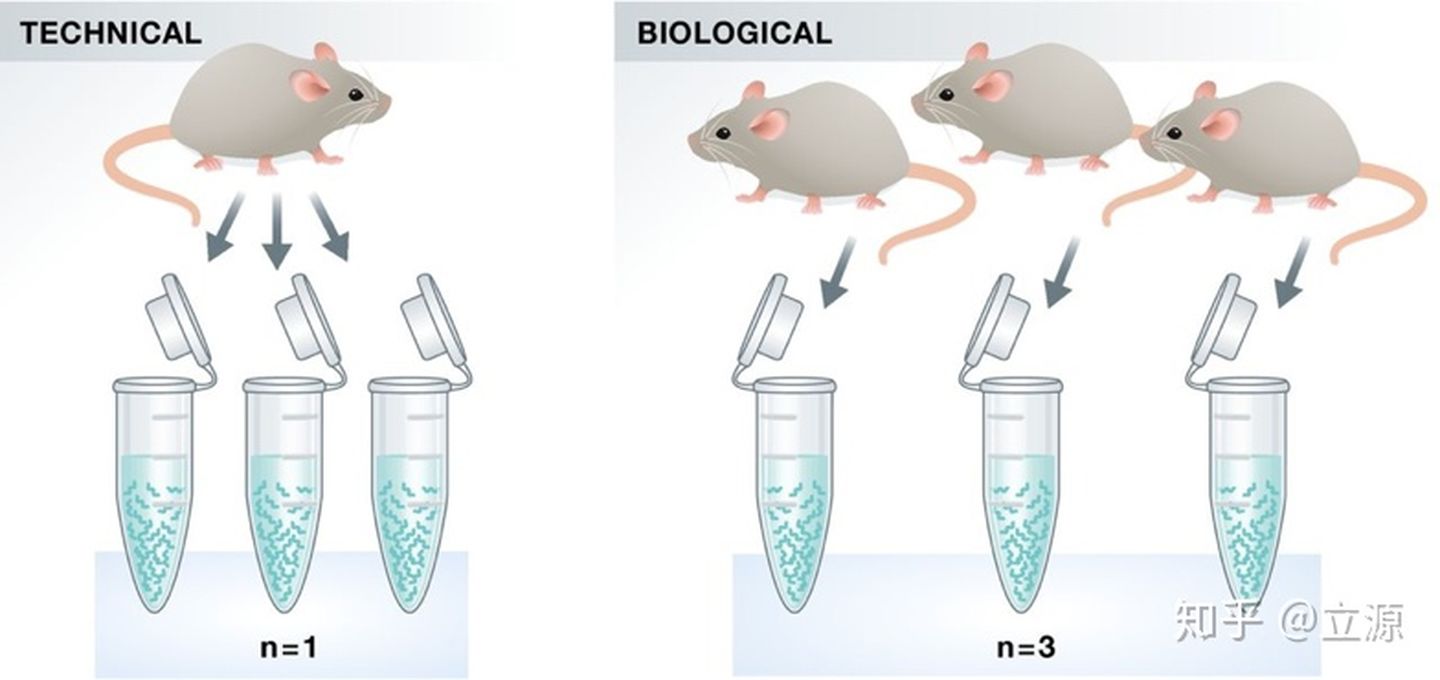
图片来源: Klaus B., EMBO J (2015) 34: 2727-2730
- 技术性重复:使用同样的生物学样本去重复技术性或实验步骤来准确衡量技术性差异并在分析中去除它
- 生物学重复:使用相同条件下的不同的生物学样本 来衡量 样本之间的生物学差异
在微阵列(microarrays)的时代,技术性重复被认为是必要的;但是,使用现在的RNA-Seq技术,技术变异比生物学变异要小得多,所以技术性重复不是必要的。
相反,对于差异表达分析来说,生物学重复是绝对必要的。对于小鼠或大鼠来说,判断是什么造成了一个不同的生物学样本或许是容易的,但对细胞系做出这种判断要困难得多。这篇文章给出了细胞系重复的一些非常好的建议。
对于差异表达分析,生物学重复越多,对生物学变异的估计要越好,我们对平均表达水平的估算也就越准确。这会使得我们数据的建模更准确,差异基因识别得越多。
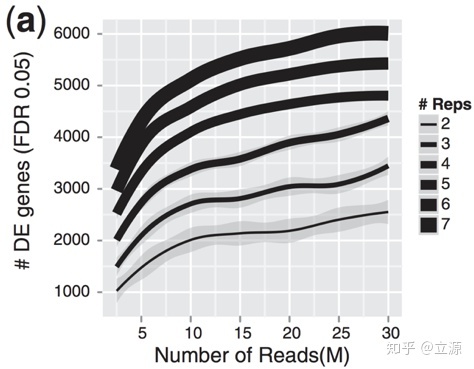
图片来源: Liu, Y., et al., Bioinformatics (2014) 30(3): 301–304
如上图所示,生物学重复比测序深度,也就是每个样本所测的reads总数要更重要。这张图片展示了测序深度和生物学重复的数目在找到的差异基因数目之间的关系。值得注意的是,有一个随着生物学重复的增加,测序深度增加,找到差异基因越多的趋势。因此,通常更多重复要好于更高的测序深度,而更高的测序深度只在检测较少表达的差异基因和进行亚型水平(isoform-level)的基因表达才是需要的。
样本混合(Sample pooling):如果可能的话,尝试避免混合个体/实验;但是,如果有绝对必要的话,然后每个样本混合集应被视作一个 单个重复(single replicate)。为了确保重复之间类似数量的变异,你将为每个混合样本集混合相同数量个体。
举个例子,如果你需要至少三个个体来获取你control重复的足够资料,至少5个个体来获得你treatment重复的足够资料,你应该聚集5个个体作control和5个个体作treatment条件。你也将确保同样条件下聚集的个体在性别,年龄等是相似的。
对于bulk RNA-Seq来说,重复几乎总是优先于更高的测序深度。但是,指导方针随着进行的实验和需要的分析而不同。我们在下面列出了一些帮助实验设计的重复和测序深度的常见准则:
- 常用的基因水平差异表达:
- ENCODE准则建议对每个样本进行30,000,000reads的单端测序
- 如果重复足够多(>3)的话,每个样本15,000,000reads通常是足够的
- 如果可能的话,把钱花在更多的生物学重复上
- 通常推荐read长度>=50 bp
- 检测低表达基因的基因水平表达差异
- 类似的,多个重复比提升测序深度要更好
- 根据表达水平,深度测序至少在30-60,000,000reads以上(如果重复够多,从30,000,000开始)
- 通常推荐read长度>=50 bp
- 亚型水平的差异表达:
- 在熟悉的亚型中,建议每个样本至少深度为30,000,000reads的双端测序
- 未知亚型应该需要更大的深度(>60,000,000 reads每个样本)
- 选择生物学重复,而不是双端/深度测序
- 通常推荐read长度>=50 bp,但是更长效果会更好,因为reads将更有可能穿过外显子连接(exon junctions)
- 对RNA质量进行仔细的质控。注意使用高质量建库方法和严格的分析来获得高RIN的样品
- 其他类型的RNA分析(intron retention, small RNA-Seq等):
- 根据分析不同,推荐不同
- 基本上,更多生物学重复总是更好的!
注:用于估算基因组测序深度的因素是“覆盖范围”- 核苷酸被测次数 "覆盖 "基因组多少次。这个指标对于基因组(全基因组测序)来说并不精确,但是它已经足够好了,并且被广泛使用。然而,这个指标对转录组并不适用,因为即使你可能知道基因组有百分之多少的转录活性,基因的表达是高度可变的。
混淆(Confounding)
一个混淆的RNA-Seq实验是指你在实验数据中无法辨别两个不同变异来源的独立效应。
例如,我们知道性别对于基因表达有很大影响,如果我们所有控制组的小鼠都是雌性而治疗组的小鼠都是雄性,那么我们的治疗效应会被性别所混淆。我们不能够从性别的影响中区分出治疗影响。
为了避免混淆:
- 如果有可能的话,确保每个条件下的动物有相同的性别,年龄和批次
- 如果不可能的话,确保在不同条件中均匀地划分动物
批次效应(Batch effects)
批次效应是RNA-Seq分析的一个重要问题。一张从Hicks SC, et al., bioRxiv (2015)中截取的图片很好地解释了这个现象:左侧描绘了实验设计,通过在每个批次中都有来自两个样本组的样本,展示了对批次的良好利用。在最右侧,绘制了一个PCA的例子,样本会按批次分离开来。说明批次对基因表达的影响通常会大于来自实验变量的影响,因此设计实验时,我们必须在统计模型中考虑到这一点。我们将在下面更详细地讨论这个问题。
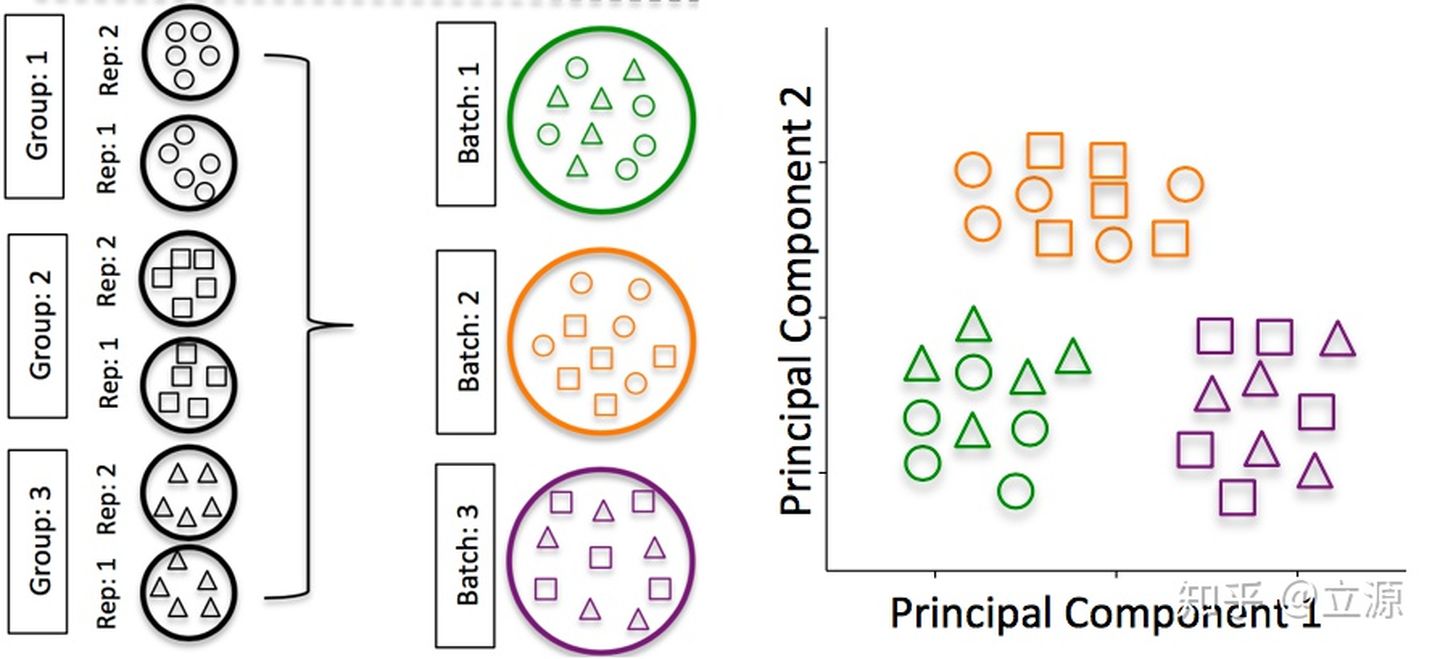
图片来源: Hicks SC, et al., bioRxiv (2015)
这些研究设计中由于批次处理不当所产生的问题在这篇文章中有着精彩的阐述。
如何知道你是否有批次问题?
- 所有的RNA提取都是在同一天进行的吗?
- 所有的建库工作都是在同一天进行的吗?
- 所有样品的RNA提取或建库是否由同一个人完成?
- 你是否对所有样品都使用了相同的试剂?
- 你是否在同一地点进行了RNA的提取或建库?
如果这些答案中有一个是“不”,那么你就有批次问题。
有关批次问题的最佳实践
- 在实验设计中尽可能地避免产生批次
- 如果无法避免批次:
- 不要混淆不同批次的实验,即不要一个批次只做一个条件下的样本:
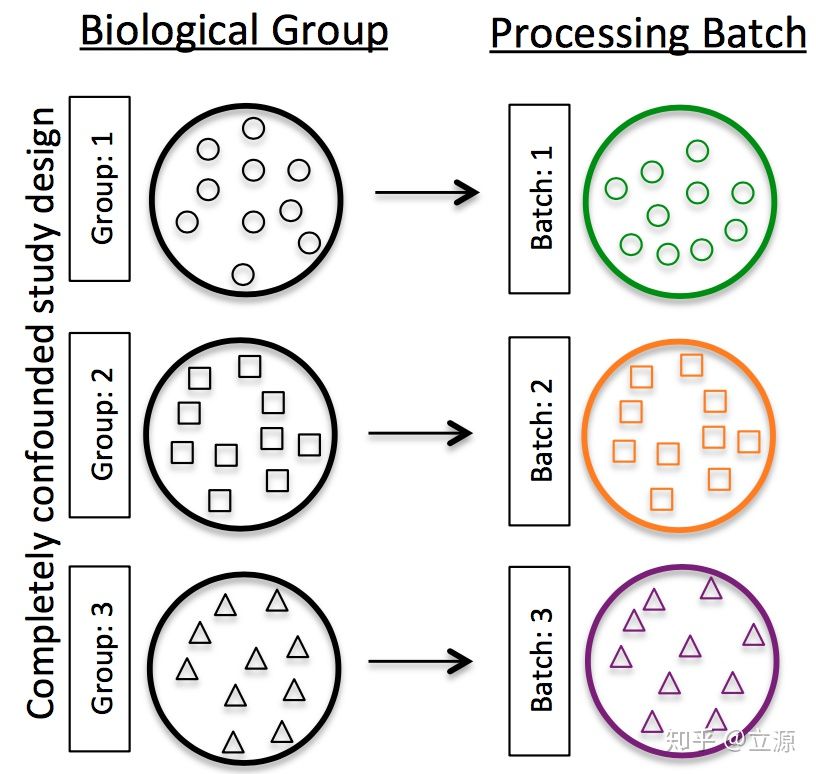
-
- 请在不同批次中做不同类别样品的重复(replicates)。如果是想找出不同处理条件下的差异基因或者在群体水平上下结论,重复自然越多越好(当然要大于2)。
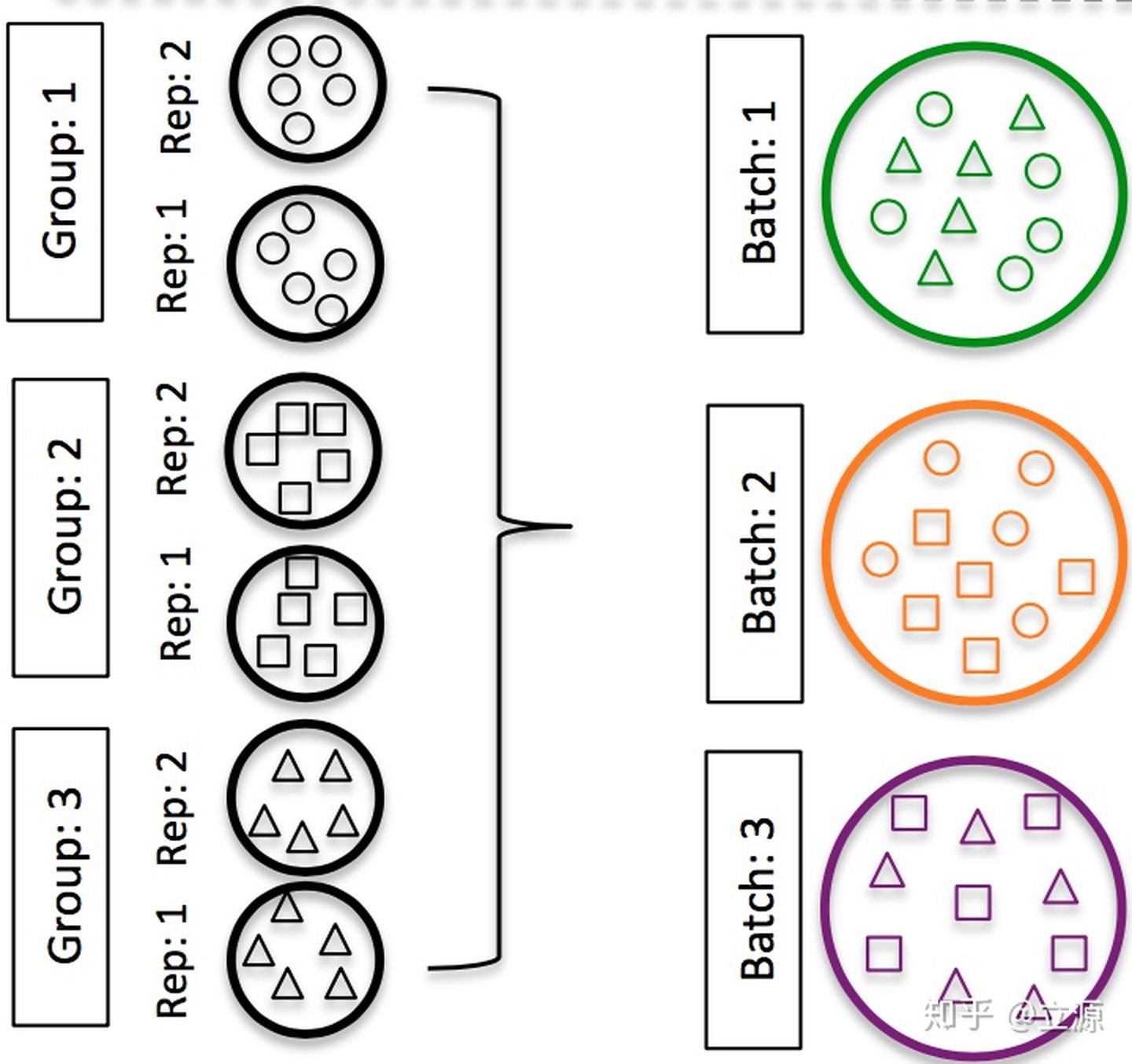
- 请在实验元数据(experimental metadata)中包含批次信息。这样在分析中我们就可以去除由于批次而产生的差异。当我们有这些信息的话就不会影响我们最终的结果。