Fastq文件大小和测序覆盖度初探
王焕威 聚道科技GeneDock 2017-04-10
(本文由GeneDock公司 Bioinformatics Engineer 王焕威撰写,转载请保留作者信息和原文链接)
引 子
在二代测序(NGS)领域中,Fastq文件大小和测序深度(即测了多少乘)是两个常用的概念,但不同人给出的Fastq文件大小与测序深度的比例可能并不一致,而且之间的关系也一直模糊不清。
故,这篇博客就试图去探讨这两者的关系及其相关概念。
基本概念
1
Fastq文件的基本格式
Fastq文件是二代测序行业中常用的原始序列文件。每4行表示一个read(测序序列),其格式示例如下:

-
第一行:为序列ID
-
第二行:序列
-
第三行:固定为“+”
-
第四行:序列的质量值(quality score)
2
Fastq文件的序列ID行
对于Fastq文件中每个序列的ID行(首行),其格式并不统一,不同来源Fastq文件的首行表示不同。
illumina测序仪的ID行一般包含测序仪、运行编号、flowcell ID、lane ID、tile ID、横纵轴坐标、索引序列等等。
(@<instrument>:<run number>:<flowcell ID>:<lane>:<tile>:<x-pos>:<y-pos> <read>:<is filtered>:<control number>:<index sequence>)
示例如下:
@EAS139:136:FC706VJ:2:5:1000:12850 1:Y:18:ATCACG
3
测序深度(coverage or depth)
测序深度或者覆盖度(coverage or depth)是指参考序列一个碱基上比对的reads的数目。计算公式为:
【测序深度 = reads长度 × 比对的reads数目 / 参考序列长度】
测序深度是NGS分析的重要质控指标。Craig Venter的文章指出全基因组应该达到30X-40X的测序深度。
* We report on the sequencing of 10,545 human genomes at 30×–40× coverage with an emphasis on quality metrics and novel variant and sequence discovery.
4
人类基因组的长度
对于人类全基因组来说,长度大约3Gbp(Giga-basepairs)。
* 3,234.83 Mb (Mega-basepairs) per haploid genome
对于人类外显子组来说,长度大约是30Mbp(Mega-basepairs)。
* The exome of the human genome consists of roughly 180,000 exons constituting about 1% of the total genome, or about 30 megabases of DNA.
另外,对于外显子组还需要考虑捕获芯片设计的问题,如Agilent和Nimblegen的不同芯片捕获区域不同。
各种对应关系
1
ASCII码和文件大小
Fastq文件所含内容均为ASCII码,每个ASCII码占用一个字节(Byte)空间。故:
Fastq文件大小 =(ID行长度 + reads长度 + 1个加号 + reads长度 + 4个换行符) × reads数目
示例如下:【计算公式为 (7 + 60 + 1 + 60 + 4) × 1 = 132B】

为了简化,我们忽略第三行的加号、换行符,并认为ID行长度在0~1个read长度,故:
【Fastq文件大小 = ~ 2.5 × reads长度 × reads数目】
2
Fastq文件大小和Fastq.gz文件大小
在传输Fastq文件过程中,经常使用gzip程序对其进行压缩,以减小文件大小,增加传输速度。而gzip对于不同Fastq文件的压缩比不同,大约在(3~5):1之间。
示例如下:【计算公式为:291852847/65573424 = 4.45078】
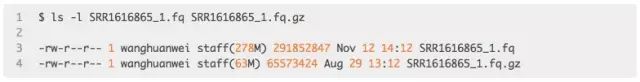
故:Fastq.gz文件大小 = ~ Fastq文件大小/4。
3
从Fastq文件到比对的reads数
由于Fastq文件经常会进行去接头等前处理的工作,比对的reads长度与原始reads长度略有不同,此处暂时忽略。
另外由于只有部分原始reads会比对(mapped)到参考基因组,因此还有一个比对率的问题。
故:比对的reads数目 = reads数目 × 比对率。比对率也是NGS分析的重要质控指标。
总 结
综合以上信息:
1. 测序深度 = reads长度 × 比对的reads数目 / 参考序列长度
2. 人类基因组 = ~3Gbp
3. Fastq文件大小 = ~ 2.5 × reads长度 × reads 数目
4. Fastq.gz文件大小=~Fastq文件大小 / 4
5. 比对的reads数目 = reads数目 × 比对率
在进行了各种近似之后(ID行的近似,参考基因组的近似,gz压缩率的近似,去接头后reads长度变化等),再假设比对率为90%,若要测30X的人类基因组需要62.5GB(Giga Base)的数据。