之前我们学习的一直都是对连续型数据的整合,其实Pathview同样也可以处理离散型数据,下面我们来看一下吧。
1. 处理离散型数据
之前我们都是在处理连续性变量,我们下面看看离散型数值要如何设置。比如我们做了两个比较组间的基因差异表达分析,将基因根据检验后的p值分为显著变化和不显著两类,构成了一组离散型数据。
我们先来构造一下离散型数据,包括基因数据和化合物的数据
gse16873.t <- apply(gse16873.d, 1, function(x) t.test(x,
alternative = "two.sided")$p.value)
sel.genes <- names(gse16873.t)[gse16873.t < 0.1]#假设p值小于0.1为显著
sel.cpds <- names(sim.cpd.data)[abs(sim.cpd.data) > 0.5]#只保留绝对值大于0.5的化合物我们设置参数discrete = list(gene = T, cpd = T)即可,这样就能画出离散型变量的通路图。图中的limit和bins参数分别是设置转换为颜色后的假值和颜色分几个区间。
pv.out <- pathview(gene.data = sel.genes,
cpd.data = sel.cpds,
pathway.id = demo.paths$sel.paths[i],
species = "hsa",
out.suffix = "sel.genes.sel.cpd",
keys.align = "y",
kegg.native = T,
key.pos = demo.paths$kpos1[i],
limit = list(gene = 5, cpd = 2),
bins = list(gene = 5, cpd = 2),
na.col = "gray",
discrete = list(gene = T, cpd = T))结果如下,虽然改变了limits和bins的值,将基因和cpd的最大值分别设置到5和2,但是分离性变量的值不变。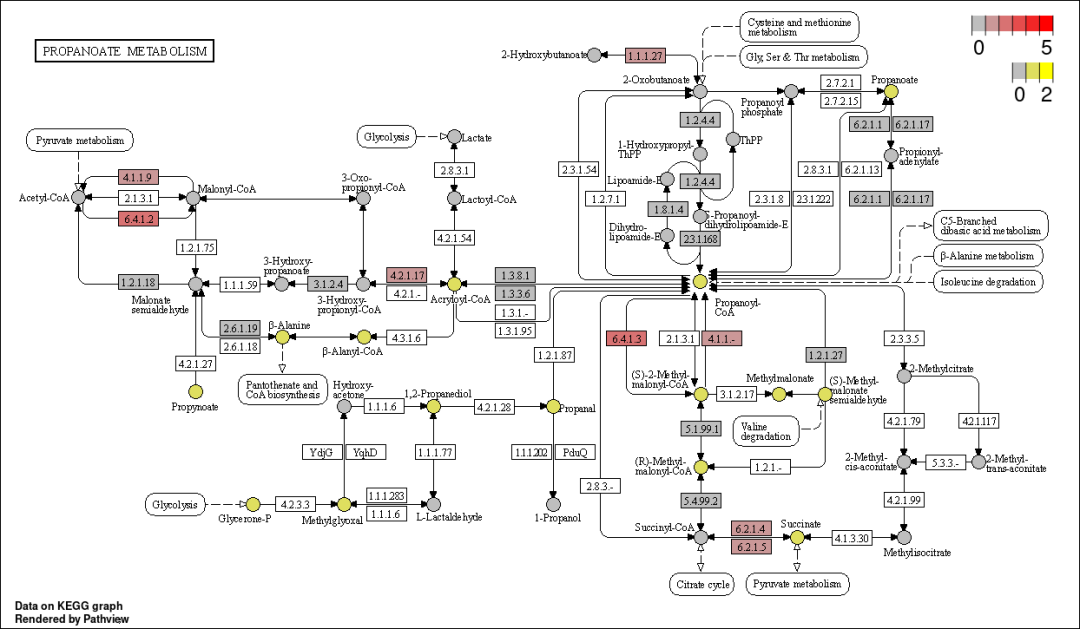
我们也可以设置基因数据为离散型,化合物为连续型变量
pv.out <- pathview(gene.data = sel.genes,
cpd.data = sim.cpd.data,
pathway.id = demo.paths$sel.paths[i],
species = "hsa",
out.suffix = "sel.genes.cpd",
keys.align = "y",
kegg.native = T,
key.pos = demo.paths$kpos1[i],
limit = list(gene = 5, cpd = 1),
bins = list(gene = 5, cpd = 10),
na.col = "gray",
discrete = list(gene = T, cpd = F))#基因数据为离散型,化合物为连续型变量结果如下: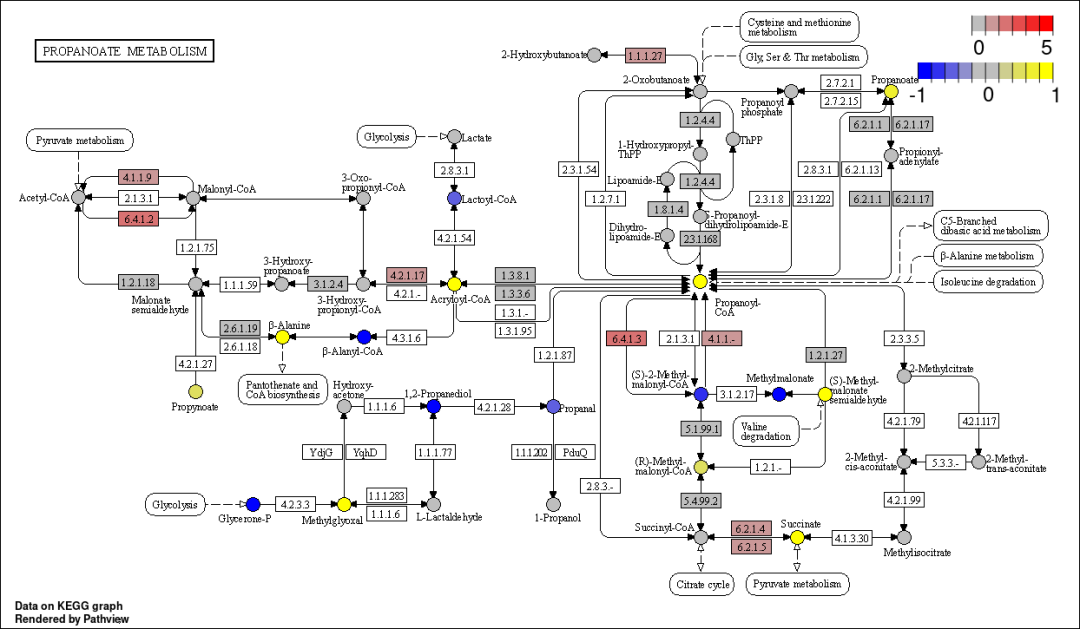
2. ID Mapping
Pathview的另一大特点就是强大的ID转换能力。Pathview中的ID转换功能包含了超过十种基因ID、蛋白质ID以及大于20中类型的化合物或代谢物ID,还包括一些额外的ID。总之,虽然使用者的数据有着不同的ID类型,但都可以准确的映射到目标KEGG通路。
此外,Pathview还能应用到超过4800种物种上,物种的类型可以通过多种形式确定,像是KEGG代码,科学名称等。本教程主要关注人类,就不过多展示其它物种的例子。
同样也是使用Pathview函数,我们只要设定参数gene.idtype和cpd.idtype为我们数据中的ID类型就好了,函数会自动匹配并转换。
需要注意的是自动转换限定于特定ID,可以查看gene.idtype.list或rn.list来确定自己的ID是否在运行使用的ID之中。
2.1可用的基因ID和化合物ID
下面展示一下Pathview中可用的基因ID以及化合物ID:
data("gene.idtype.list")
data("rn.list")
gene.idtype.list
names(rn.list)
> gene.idtype.list
[1] "SYMBOL" "GENENAME" "ENSEMBL" "ENSEMBLPROT" "UNIGENE" "UNIPROT"
[7] "ACCNUM" "ENSEMBLTRANS" "REFSEQ" "ENZYME" "TAIR" "PROSITE"
[13] "ORF"
> names(rn.list)
[1] "Agricola citation" "Beilstein Registry Number" "CAS Registry Number"
[4] "COMe accession" "ChEMBL COMPOUND" "DrugBank accession"
[7] "Gmelin Registry Number" "HMDB accession" "KEGG COMPOUND accession"
[10] "KEGG DRUG accession" "KEGG GLYCAN accession" "LIPID MAPS instance accession"
[13] "MetaCyc accession" "MolBase accession" "PDB accession"
[16] "PDBeChem accession" "Patent accession" "PubMed citation"
[19] "Reaxys Registry Number" "UM-BBD compID" "Wikipedia accession"2.2 实例展示
下面看一个不同类型ID转换并成功映射的实例:
首先先构建一个模拟单样本的化合物数据和基因表达量数据
化合物数据使用的ID是
cpd.simtypes[2],也就是"CAS Registry Number"基因数据使用的ID是
gene.idtype.list[4],也就是"ENSEMBL"需要注意的是
sim.mol.data()函数在ID数量有限时只能产生上限数值的数据
cpd.cas <- sim.mol.data(mol.type = "cpd",
id.type = cpd.simtypes[2],
nmol = 10000)
gene.ensprot <- sim.mol.data(mol.type = "gene",
id.type = gene.idtype.list[4],
nmol = 50000)
Note: "ENSEMBLPROT" has only 21768 unique IDs!
Note: "CAS Registry Number" has only 4643 unique IDs!模拟好数据后,直接调整pathview中的参数即可:
设定参数
gene.idtype和cpd.idtype为我们模拟数据时用的ID后,函数就会帮我们转换数据为KEGG ID运行过程中会有这样一句提醒
Note: multiple compounds may map to a input ID, only the first one kept!。当多个化合物对应一个ID时,只有一个会被保留。
pv.out <- pathview(gene.data = gene.ensprot, #基因数据
cpd.data = cpd.cas, #化合物数据
gene.idtype = gene.idtype.list[4], #基因使用的ID
cpd.idtype = cpd.simtypes[2], #化合物使用的ID
pathway.id = demo.paths$sel.paths[3],#通路ID
species = "hsa",
same.layer = T,
out.suffix = "gene.ensprot.cpd.cas",
keys.align = "y",
kegg.native = T,
key.pos = demo.paths$kpos2[3],#标签的位置
sign.pos = demo.paths$spos[3],#图例的位置
limit = list(gene = 3, cpd = 3),
bins = list(gene = 6, cpd = 6))结果如下: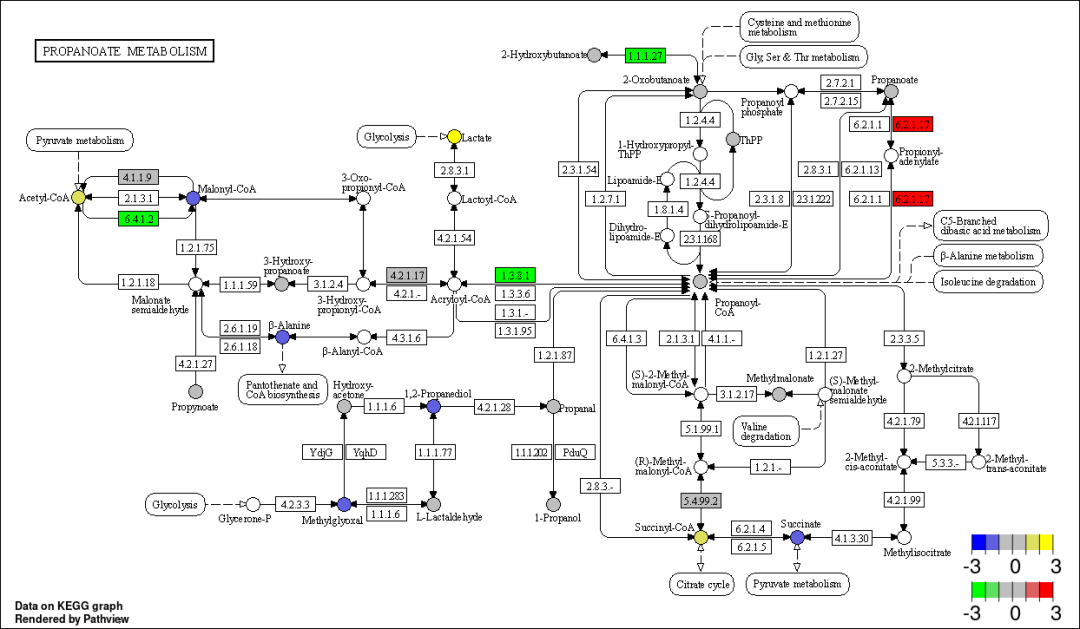
2.3外部ID的转换
对于那些不在Pathview中的外部ID,即gene.idtype.list或rn.list中不存在的ID,可以利用mol.sum()函数来转换ID。除了我们的原始数据矩阵mol.data外,我们只需要提供一个额外的id.map矩阵即可,即外部ID和KEGG标准ID间的映射矩阵。然后该函数就可以将我们数据的ID转换。
mol.sum(
mol.data,
id.map)可以利用像是id2eg()或cpdidmap()函数来产生映射矩阵。这些函数也可以独立使用。
下面举一个例子:
我们分别用
cpdidmap()和id2eg()这两个函数将上一步模拟的数据的ID做一个转换,变为标准的KEGG ID,然后合并出各自的id.map矩阵再用
mol.sum函数将模拟数据的ID转换
id.map.cas <- cpdidmap(in.ids = names(cpd.cas), #当前化合物的名称
in.type = cpd.simtypes[2],#化合物名称所属的类别,这里为"CAS Registry Number"
out.type = "KEGG COMPOUND accession")#转换的类别,转换成标志的KEGG ID
#看一下cpd的ID map矩阵,无法对应到KEGG ID的化合物名称用NA表示
> head(id.map.cas)
CAS Registry Number KEGG COMPOUND accession
[1,] "128470-16-6" NA
[2,] "465-42-9" "C08584"
[3,] "25812-30-0" NA
[4,] "4790-08-3" "C04185"
[5,] "95789-30-3" NA
[6,] "138-14-7" NA
#用mol.sum函数将原始数据做一个筛选和ID转换
cpd.kc <- mol.sum(mol.data = cpd.cas,
id.map = id.map.cas) #根据制作好的外部ID和KEGG ID间的映射,对数据的ID做一个转换
#对基因的外部ID转换也是相同的方法,不过用到的是id2eg函数
id.map.ensprot <- id2eg(ids = names(gene.ensprot),
category = gene.idtype.list[4],
org = "Hs")
gene.entrez <- mol.sum(mol.data = gene.ensprot,
id.map = id.map.ensprot)最后再使用pathview函数即可,画出的图像与上方一致:
pv.out <- pathview(gene.data = gene.entrez,
cpd.data = cpd.kc,
pathway.id = demo.paths$sel.paths[i],
species = "hsa",
same.layer = T,
out.suffix = "gene.entrez.cpd.kc",
keys.align = "y",
kegg.native = T,
key.pos = demo.paths$kpos2[i],
sign.pos = demo.paths$spos[i],
limit = list(gene = 3, cpd = 3),
bins = list(gene = 6, cpd = 6))3. 网页版本
Pathway的作者不仅提供了R包,同时也创建了一个网页应用,如果暂时不熟悉R语言,可以去网页上试着使用Pathway。
网址如下:
https://pathview.uncc.edu/
4. 小结
今天我们学习了如何用Pathview包处理离散型数据。此外还学习了包中强大的ID转换功能,包括内部已有的ID的转换,和外部ID的转换方法。到目前我们算是对Pathview包有了一个系统的了解。下一期我们来看一下高分文章中都是如何使用这个包,又能有怎样的新花样。