前面几期“干货预警——原来基因功能富集分析这么简单!”、“【R语言】——基因GO/KEGG功能富集结果可视化(保姆级教程)”和“【R语言】——基因GO/KEGG功能富集分析!超级简单的保姆级教程!”分别介绍了如何使用DAVID在线分析工具对基因进行GO/KEGG功能富集分析、使用R ggplot包对获得的基因GO/KEGG功能富集结果进行可视化、使用R clusterProfiler包和R AnnotationHub包对基因进行GO/KEGG功能富集分析、OrgDb包制作以及结果可视化等。本期主要重新归纳和总结了富集结果可视化的代码。
有同学反映前面几期富集分析结果可视化有点儿乱,可以归纳总结下。故,我重新整理了一波,并补充了可对多个分组的富集结果进行可视化以及富集结果绘制圈图等的代码。希望可以帮助到大家。
一、单组富集结果可视化
(柱状图+气泡图)
1 数据准备
数据输入格式(xlsx格式):
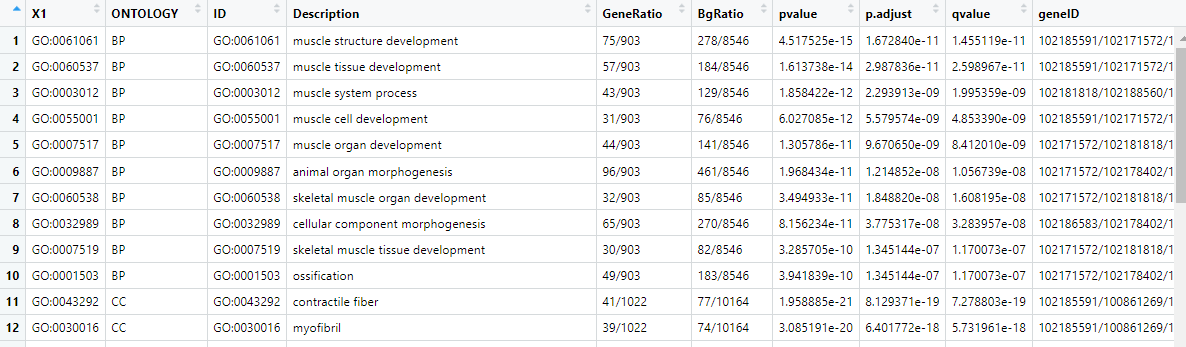
2 R包加载、数据导入及处理
#下载包#
install.packages("ggplot2")
install.packages("openxlsx")
#加载包#
library(ggplot2)
library(openxlsx)
#导入数据#
remove(list = ls()) #清除 Global Environment
getwd() #查看当前工作路径
setwd("C:/Rdata/jc") #设置需要的工作路径
list.files() #查看当前工作目录下的文件
go_enrich = read.xlsx("enrich-gene.xlsx",sheet= "ONTOLOGY",sep=',')
head(go_enrich)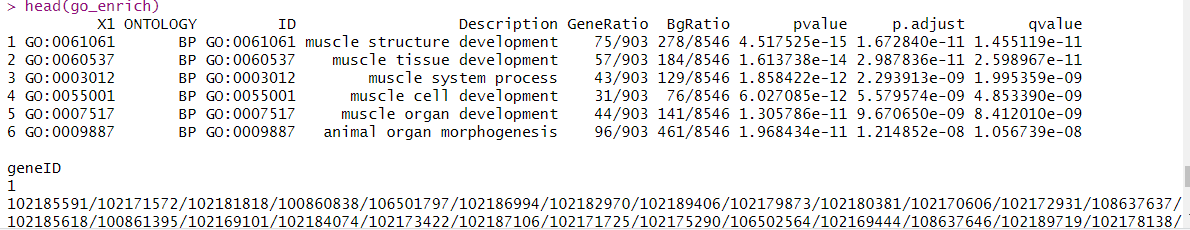
#数据处理#
go_enrich$term <- paste(go_enrich$ID, go_enrich$Description, sep = ': ') #将ID与Description合并成新的一列
go_enrich$term <- factor(go_enrich$term, levels = go_enrich$term,ordered = T) #转成因子,防止重新排列3 富集结果可视化
#纵向柱状图-根据ONTOLOGY类型绘制#
p1 <- ggplot(go_enrich,
aes(x=term,y=Count, fill=ONTOLOGY)) + #x、y轴定义;根据ONTOLOGY填充颜色
geom_bar(stat="identity", width=0.8) + #柱状图宽度
scale_fill_manual(values = c("#6666FF", "#33CC33", "#FF6666") ) + #柱状图填充颜色
coord_flip() + #让柱状图变为纵向
xlab("GO term") + #x轴标签
ylab("Gene Number") + #y轴标签
labs(title = "GO Terms Enrich")+ #设置标题
theme_bw()
p1
#根据ONTOLOGY分类信息添加分组框#
p1+facet_grid(ONTOLOGY~., scale = 'free_y', space = 'free_y')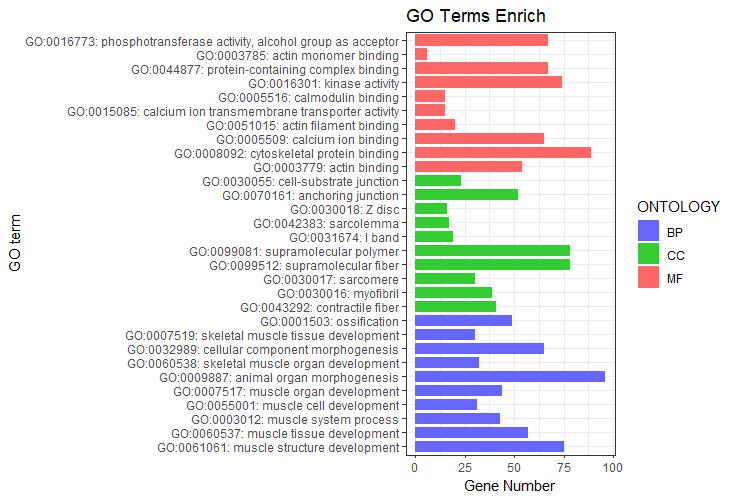
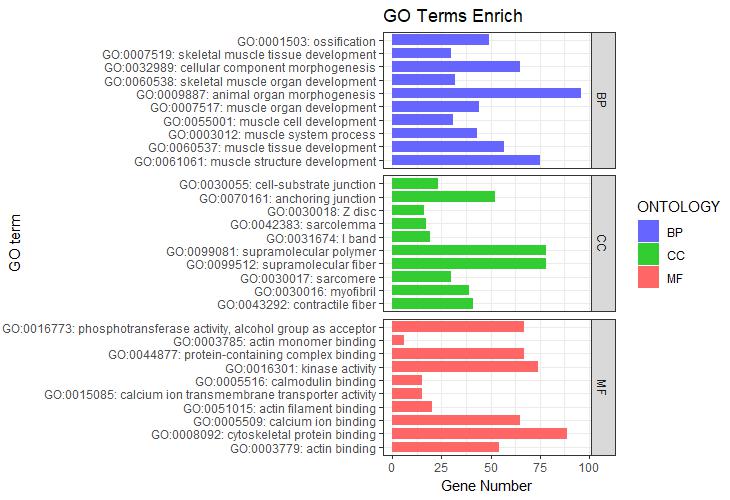
图1 纵向柱状图
#横向柱状图-根据ONTOLOGY类型绘制#
p2 <- ggplot(go_enrich,
aes(x=term,y=Count, fill=ONTOLOGY)) + #x、y轴定义;根据ONTOLOGY填充颜色
geom_bar(stat="identity", width=0.8) + #柱状图宽度
scale_fill_manual(values = c("#6666FF", "#33CC33", "#FF6666") ) + #柱状图填充颜色
xlab("GO term") + #x轴标签
ylab("Gene Number") + #y轴标签
labs(title = "GO Terms Enrich")+ #设置标题
theme_bw() +
theme(axis.text.x=element_text(family="sans",face = "bold", color="gray50",angle = 70,vjust = 1, hjust = 1 )) #对字体样式、颜色、还有横坐标角度()
p2
#根据ONTOLOGY分类信息添加分组框#
p2 + facet_grid(.~ONTOLOGY, scale = 'free_x', space = 'free_x')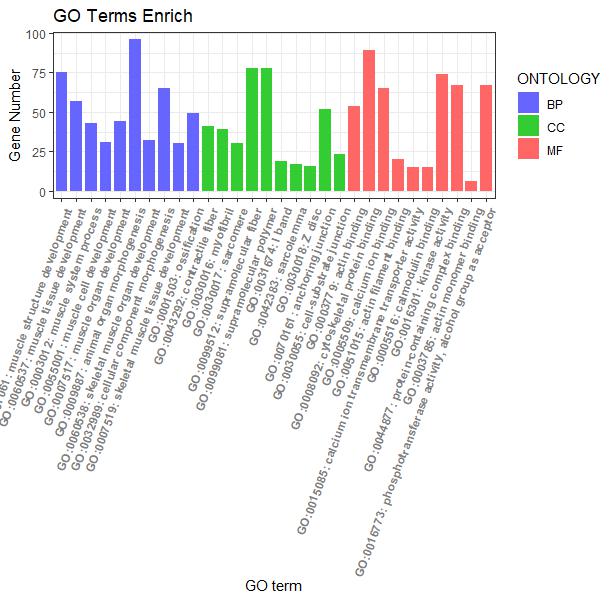
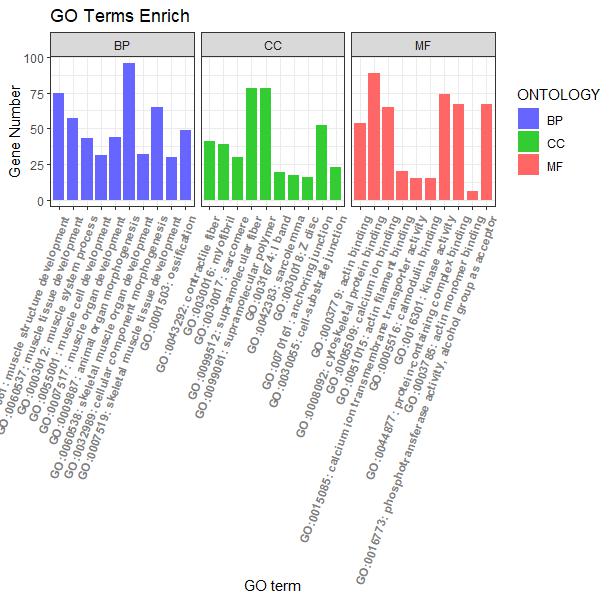
图2 横向柱状图
#纵向柱状图——-根据pvalue值绘制#
p3 <- ggplot(go_enrich,aes(y=term,x=Count,fill=pvalue))+ #x、y轴定义;根据pvalue填充颜色
geom_bar(stat = "identity",width=0.8)+ #柱状图宽度设置
scale_fill_gradient(low = "red",high ="blue" )+
labs(title = "GO Terms Enrich", #设置标题、x轴和Y轴名称
x = "Gene number",
y = "GO Terms")+
theme(axis.title.x = element_text(face = "bold",size = 16),
axis.title.y = element_text(face = "bold",size = 16),
legend.title = element_text(face = "bold",size = 16))+
theme_bw()
p3
#根据ONTOLOGY分类信息添加分组框#
p3+facet_grid(ONTOLOGY~., scale = 'free_y', space = 'free_y')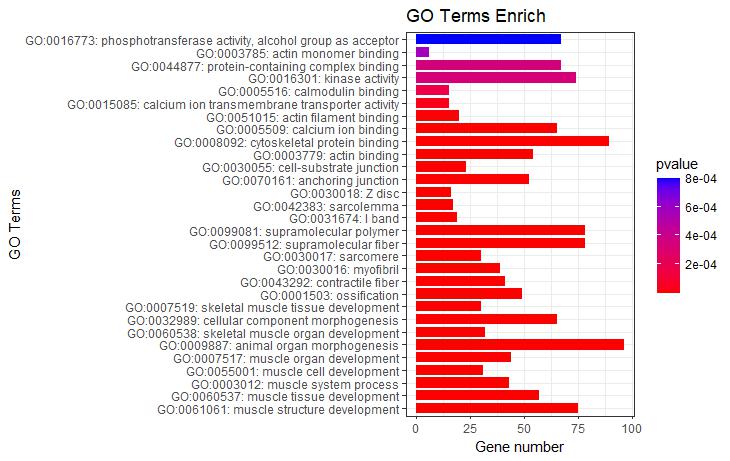
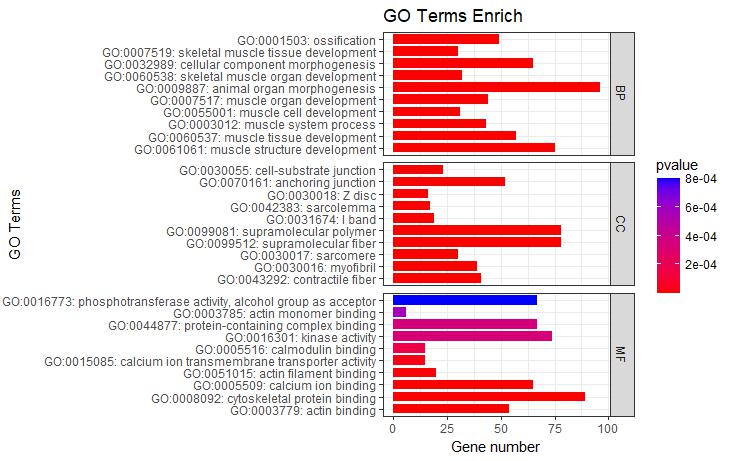
图3 根据pvalue值绘制纵向柱状图
#横向柱状图-根据pvalue值绘制#
p4 <- ggplot(go_enrich,
aes(x=term,y=Count, fill=pvalue)) + #x、y轴定义;根据ONTOLOGY填充颜色
geom_bar(stat="identity", width=0.8) + #柱状图宽度
scale_fill_gradient(low = "red",high ="blue" ) + #柱状图填充颜色
xlab("GO term") + #x轴标签
ylab("Gene Number") + #y轴标签
labs(title = "GO Terms Enrich")+ #设置标题
theme_bw() +
theme(axis.text.x=element_text(family="sans",face = "bold", color="gray50",angle = 70,vjust = 1, hjust = 1 )) #对字体样式、颜色、还有横坐标角度()
p4
#根据GO富集分析分类信息添加分组框#
p4 + facet_grid(.~ONTOLOGY, scale = 'free_x', space = 'free_x')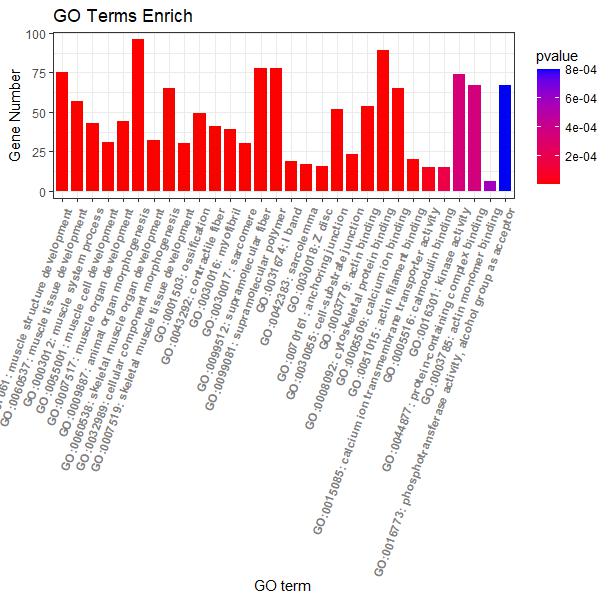
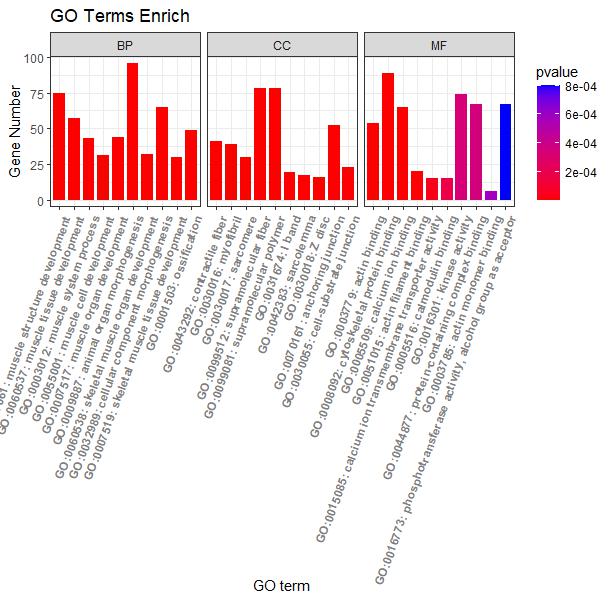
图4 pvalue值绘制的横向柱状图
#气泡图#
P5 <- ggplot(go_enrich,
aes(y=term,x=Count))+
geom_point(aes(size=Count,color=pvalue))+
scale_color_gradient(low = "red",high ="blue")+
labs(color=expression(PValue,size="Count"),
x="Gene Ratio",y="GO term",title="GO Enrichment")+
theme_bw()
P5
#根据ONTOLOGY分类信息添加分组框#
P5 + facet_grid(ONTOLOGY~., scale = 'free_y', space = 'free_y')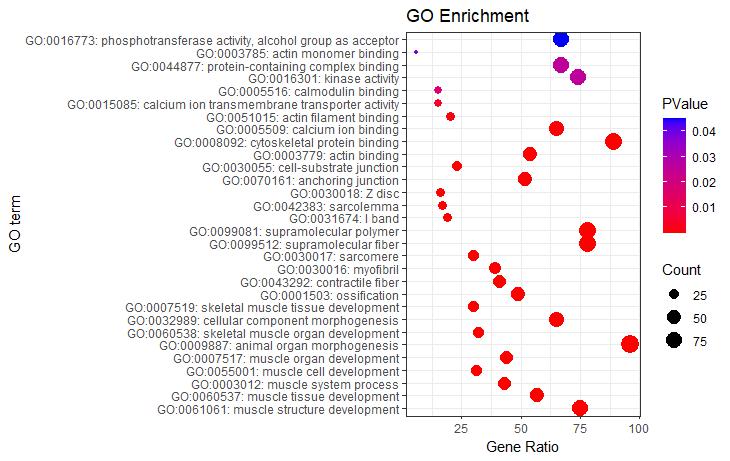
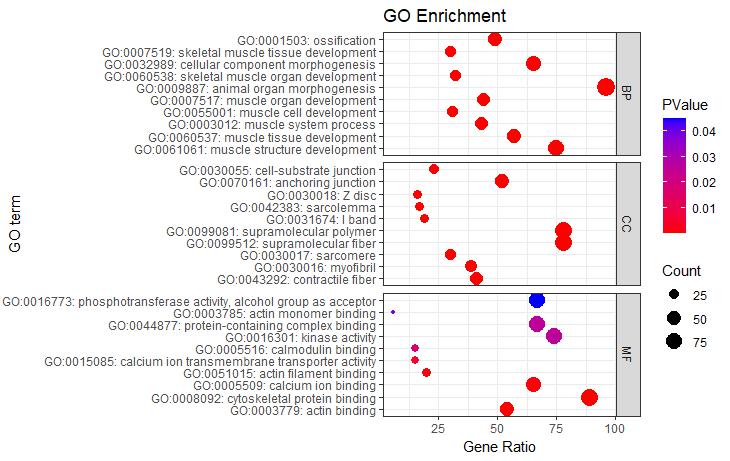
图5气泡图
二、多组富集结果可视化
(气泡图)
1 数据准备
数据输入格式(xlsx格式):
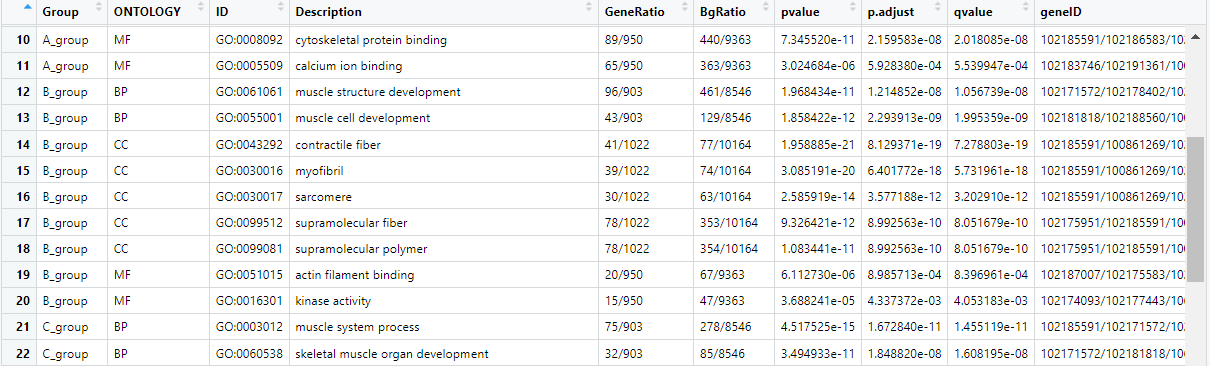
2 数据导入、处理及可视化
#气泡图——多组数据#
go_enrich = read.xlsx("enrich-gene.xlsx",sheet= "多组GO",sep=',')
head(go_enrich)
go_enrich$term <- paste(go_enrich$ID, go_enrich$Description, sep = ': ') #将ID与Description合并成新的一列
#绘图#
P6 <- ggplot(go_enrich,
aes(y=term,x=Group))+
geom_point(aes(size=Count,color=pvalue))+ #Count表示气泡图中点的大小,并按pvalue值进行着色
scale_color_gradient(low = "red",high ="blue")+
labs(color=expression(PValue,size="Count"),
x="Group",y="GO term",title="GO Enrichment")+
theme_bw()
P6
#根据ONTOLOGY分类信息添加分组框#
P6 + facet_grid(ONTOLOGY~., scale = 'free_y', space = 'free_y')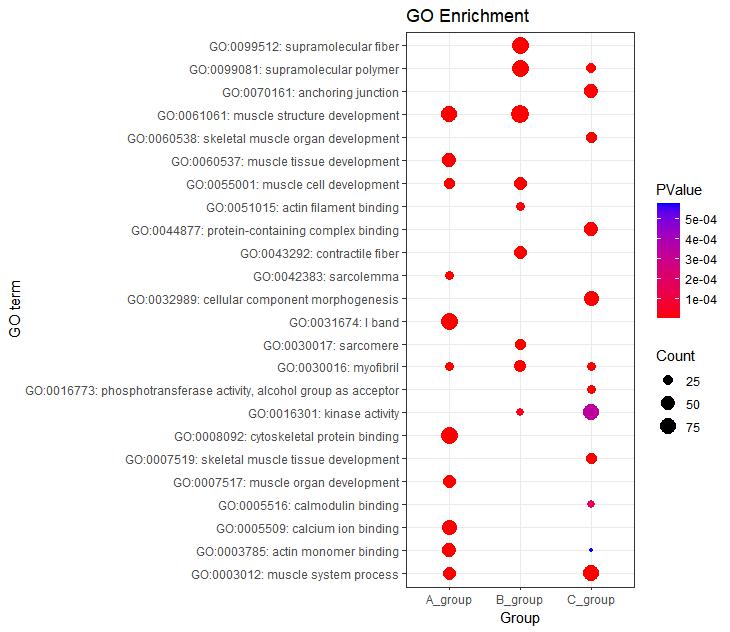
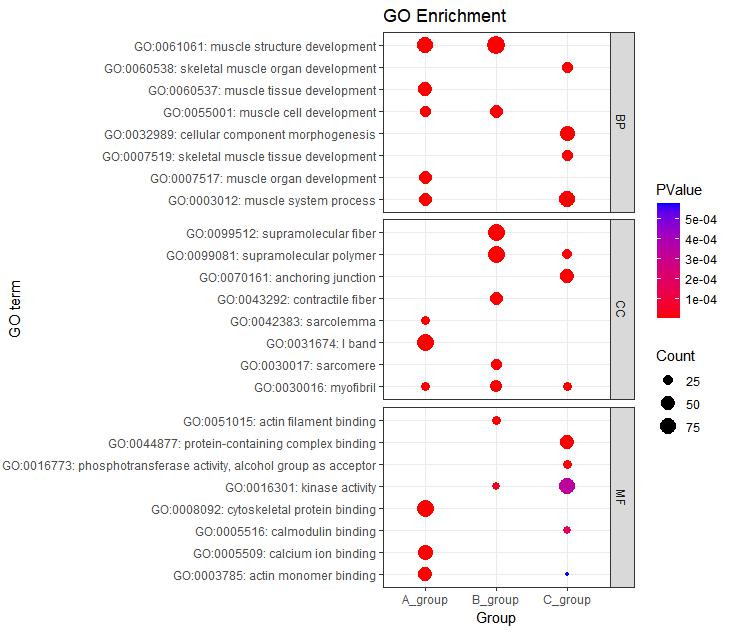
图6 多组气泡图
三、单组富集结果可视化
(圆圈图)
1 数据准备
数据输入格式(xlsx格式):
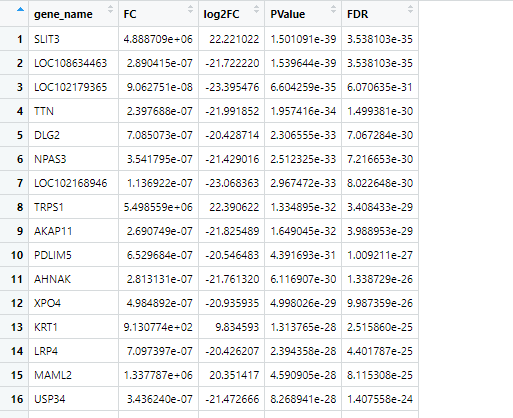
注:“ego”所载入的数据格式
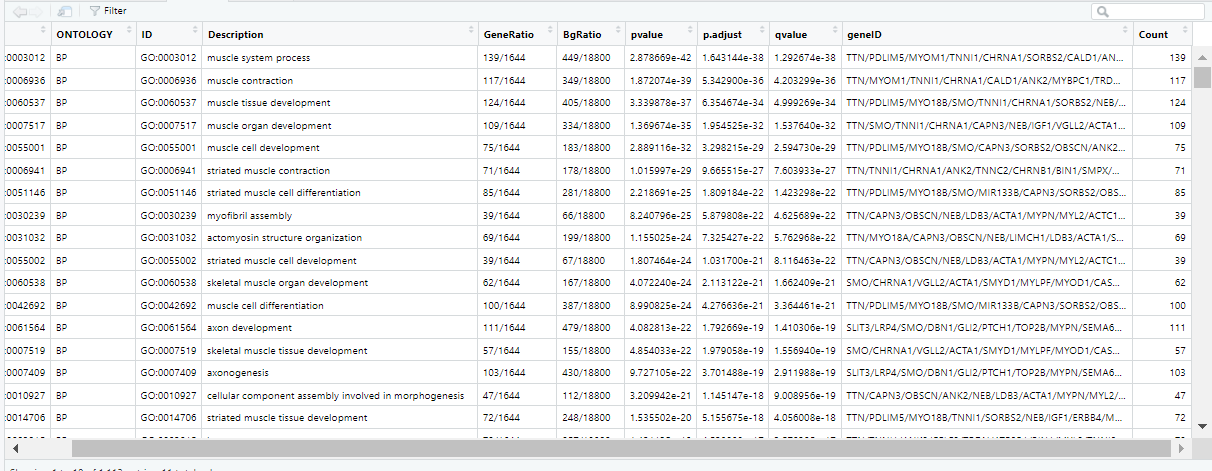
注:“GO”所载入的数据格式
2 R包加载、数据导入及处理
#功能富集圈图#
#包下载与加载#
install.packages("stringi")
install.packages("GOplot")
library(stringi) #处理表格数据的包
library(GOplot)
#数据载入#
ego = read.xlsx("enrich-gene.xlsx",sheet= "enrich_genes",sep=',')
GO = read.xlsx("enrich-gene.xlsx",sheet= "ONTOLOGY_全",sep=',')
# 构建数据框矩阵
go=data.frame(Category = GO$ONTOLOGY,ID = GO$ID,Term = GO$Description, Genes = gsub("/", ", ", GO$geneID), adj_pval = GO$p.adjust)
# 构建数据框矩阵
genelist=data.frame(ID = ego$gene_name, logFC = ego$log2FC)
row.names(genelist)=genelist[,1] #行命名
circ <- circle_dat(go, genelist)
termNum = 5 #限定term数目
termNum=ifelse(nrow(go)<termNum,nrow(go),termNum)
geneNum = nrow(genelist) #限定基因数目可以改为数字3 圈图绘制
# 差异圈图
chord <- chord_dat(circ, genelist[1:geneNum,], go$Term[1:termNum])
#pdf(file="GOcircos.pdf",width = 10,height = 10.2) #PDF格式下载
p7 <- GOChord(chord,
space = 0.001, # 基因之间的间距
gene.order = 'log2FC', # 排序基因
gene.space = 0.2, # 基因离圆圈距离
gene.size = 1, # 基因字体大小
border.size = 0.1, # 线的大小
process.label = 7) # GO名称大小
p7
dev.off()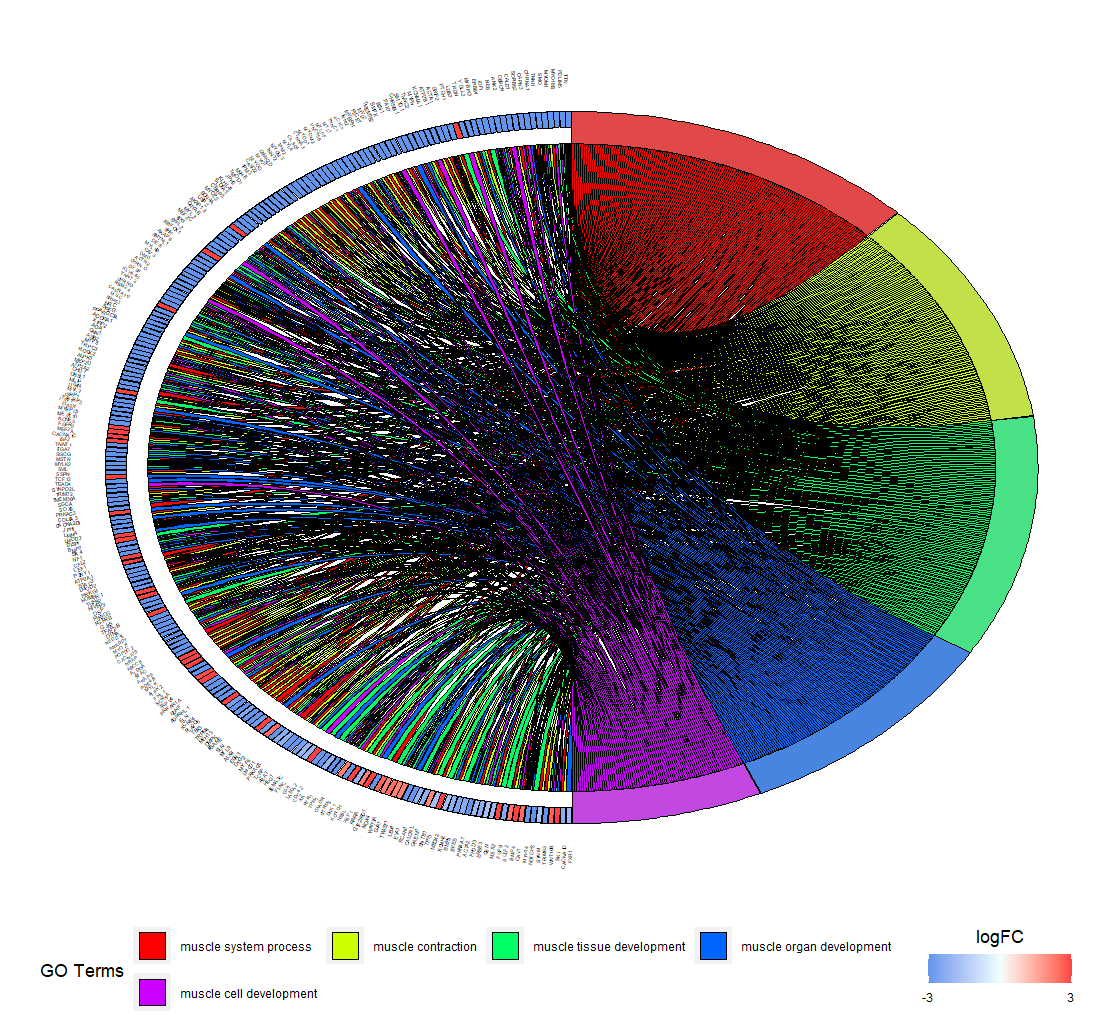
图7 富集结果圈图
好了本次分享就到这里,下期将有更精彩内容,敬请期待。

关注“在打豆豆的小潘学长”公众号,发送“富集分析3”获得完整代码包和演示数据。