轨迹分析是单细胞测序分析中重要的组成部分,它基于细胞谱系之间“具有中间态细胞”的理论基础,通过结合先验知识(细胞注释、markers)、细胞基因表达改变等,为在单细胞测序数据赋予了“假时间”(pseudotime)这一重要概念。基于pseudotime提供的时间维度,研究者得以进一步揭示细胞表型(phenotype)改变的连续过程,推断其中的基因调控机制。此前我们曾经写过几篇关于轨迹分析的推文,微信公众号:生信小博士
在pseudotime的概念之上,进一步通过算法衍生了“细胞分化潜能”或者“细胞干性”(Cell fate probabilities)等符合生物学常识的概念。例如,2020年发表在Science的CytoTRACE是一个简单但强有力的算法,它是基于每个细胞表达的基因数量,并利用这一转录多样性的度量来开发的计算框架。但是CytoTRACE也有一定的局限性,首先它是基于无监督的算法,非人为指定起点或终点,这就会导致算法出现错误的可能性,比如时序倒置,或一些显著背离生物学发展轨迹的方向。
说个题外话。有同学可能会提到,无监督的算法是否更能反映真实的改变情况,如果人为指定会不会出现过度干预或操纵的可能性。诚然,每一种算法或实验都有其局限性,但是目前关于有监督或无监督算法在单细胞各个研究领域中的应用都存在争议。就以最常见的降维分群注释为例,我们会看到越来越多基于有监督或半监督的聚类算法大放异彩,比如scANVI,以及各种单细胞图谱,他们的目的都是为了提供预先注释的信息以获得更加准确的注释。当然我个人并不懂算法开发,但先验知识与算法的有机结合呈现的优势无疑是巨大的,它能够避免一些无谓的、甚至离谱的错误出现。

今天我们分享另一个基于Python的工具Palantir。该方法于近年发表在Nature Biotechnology上。与CytoTRACE不同,Palantir基于随机漫步的算法,将细胞命运视为概率事件来模拟不同分化轨迹,并利用熵来衡量轨迹上细胞的可塑性,从而生成高分辨率的假时间排序,并为每个细胞状态分配了分化到不同终端状态的概率(Cell fate probabilities)。
环境配置
创建一个新的虚拟环境
conda create -n palantir python=3.8 -y
conda activate palantir
安装所依赖的python包,这里需要注意,palantir的最新版(1.3.1)可能会出现环境或配置错误,建议安装1.0.1版本。
pip install PhenoGraph
pip install rpy2
pip install palantir==1.0.1
配置kernel。
pip install ipykernel
# 配置jupyterlab内核
python -m ipykernel install --user --name=palantir --display-name palantir
加载Python包和全局设置
import palantir
import scanpy as sc
import numpy as np
import pandas as pd
import os
# Plotting
import matplotlib
import matplotlib.pyplot as plt
import seaborn as sns
import warnings
# Inline plotting
%matplotlib inline
sns.set_style('ticks')
matplotlib.rcParams['figure.figsize'] = [4, 4]
matplotlib.rcParams['figure.dpi'] = 100
matplotlib.rcParams['image.cmap'] = 'Spectral_r'
warnings.filterwarnings(action="ignore", module="matplotlib", message="findfont")
# Reset random seed
np.random.seed(5)
# 更换到相应的项目路径
os.chdir('path/to/project')
数据准备
官方提供的数据下载方式:
# Load sample data
data_dir = os.path.expanduser("./")
download_url = "https://dp-lab-data-public.s3.amazonaws.com/palantir/marrow_sample_scseq_counts.h5ad"
file_path = os.path.join(data_dir, "marrow_sample_scseq_counts.h5ad")
ad = sc.read(file_path, backup_url=download_url)
ad
# AnnData object with n_obs × n_vars = 4142 × 16106
如果你加载的是本地数据,或是整合的数据,需要明确的是:数据必须无批次信息或经过批次矫正。本次官方提供的数据只有一个样品,不存在批次效应。如果你用的是自己的数据,一定要系统评估批次效应。你也可以将上面的链接复制到浏览器窗口中下载。然后加载数据:
adata = sc.read_h5ad('marrow_sample_scseq_counts.h5ad')
adata
# AnnData object with n_obs × n_vars = 4142 × 16106
数据预处理:
# 计算线粒体基因比例
adata.var['mt'] = adata.var_names.str.startswith('MT-')
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
# 简单过滤
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
adata = adata[adata.obs.n_genes_by_counts < 4000, :]
adata = adata[adata.obs.pct_counts_mt < 5, :]
# 高变基因
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
# 归一化
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
sc.pp.scale(adata, max_value=10)
# PCA
# 如果你的是整合数据,应该使用 corrected PCA embedding
sc.tl.pca(adata, svd_solver='arpack')
sc.pl.pca(adata, color='CD34')
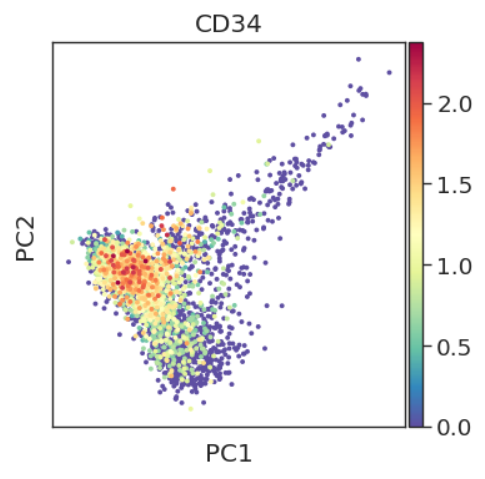
根据Palantir官方的细胞注释信息,我们对细胞进行注释
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata)
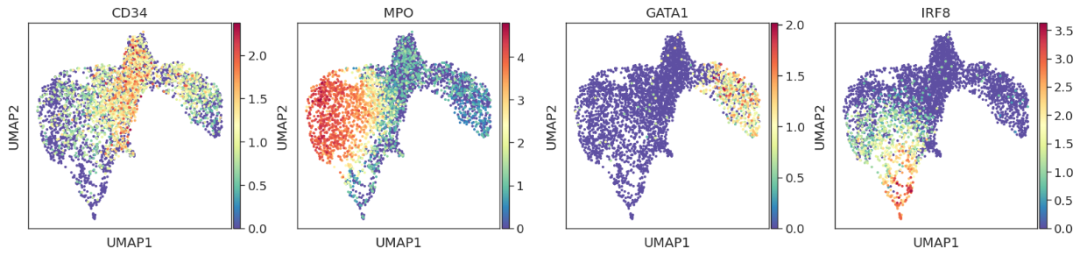
sc.pl.umap(adata, color=["CD34", "MPO", "GATA1", "IRF8"])
细胞重命名
sc.tl.leiden(adata, resolution=0.5)
sc.pl.umap(adata, color='leiden') # 图就不展示了
# 这里我们使用字典进行重命名,以防止错乱,尤其是你的分类特别多的时候
# scanpy提供的解决方案是 adata.rename_categories()
celltype_dict = {
'0':'HSC',
'1':'Ery',
'2':'Mono',
'3':'Mono',
'4':'Mono',
'5':'DC',
'6':'DC'
}
adata_rename = adata.copy()
adata_rename.obs['CellType'] = adata_rename.obs['leiden']
# 重命名
new_celltype = []
for subtype in adata_rename.obs['CellType']:
new_subtype = celltype_dict.get(subtype, subtype)
new_celltype.append(new_subtype)
adata_rename.obs['CellType'] = pd.Categorical(new_celltype)
sc.pl.umap(adata_rename, color='CellType', frameon=False)

Diffusion Map和Force Directed Graph
可能很多做单细胞的小伙伴都没有接触过Diffusion Map(DM)和Force Directed Graph(FDG)这两个概念。这其中重要原因是Seurat和Scanpy提供在标准分析流程没有提及(实际上Scanpy有内置相应的函数)。这其实是非常重要的两个概念,很多高分文章的拟时序分析都应用了这两种降维方式。
Diffusion Map(扩散映射)和force directed graph(力导向图)是在单细胞研究中常用的数据可视化和降维方法。Diffusion Map是一种基于扩散过程的降维方法。它通过计算数据点之间的相似性和距离,构建一个扩散矩阵,然后对该矩阵进行特征值分解,得到数据的低维表示。Diffusion Map可以帮助发现数据中的潜在结构和关系,尤其适用于非线性数据。Force directed graph是一种可视化方法,用于展示数据点之间的关系和相互作用。在force directed graph中,数据点被表示为节点,而数据点之间的关系则通过边来表示。边的长度和弹簧力的大小根据数据点之间的相似性和距离进行调整,从而使得相似的数据点更加靠近,不相似的数据点则远离。通过调整边的长度和力的大小,可以得到一个具有良好可视化效果的图形。
在单细胞研究中,DM和FDG可以用于数据的可视化和降维。它们可以帮助研究人员理解和解释单细胞数据的结构和关系。通过应用DM,可以将高维的单细胞数据降低到较低的维度,从而更好地展示数据的特征和变化。而FDG可以将单细胞数据的相似性和关系以图形的形式展示出来,从而帮助研究人员发现细胞之间的相互作用和群落结构。
我们通过这个实例来看看他们有什么不同,以及为什么这两种降维方式适合做轨迹推断:
adata = adata_rename.copy()
# diffmap
sc.pp.neighbors(adata, n_neighbors=20, n_pcs=50, use_rep='X_pca', method='gauss')
sc.tl.diffmap(adata)
sc.pl.embedding(adata,'diffmap',color=['CellType','leiden'])
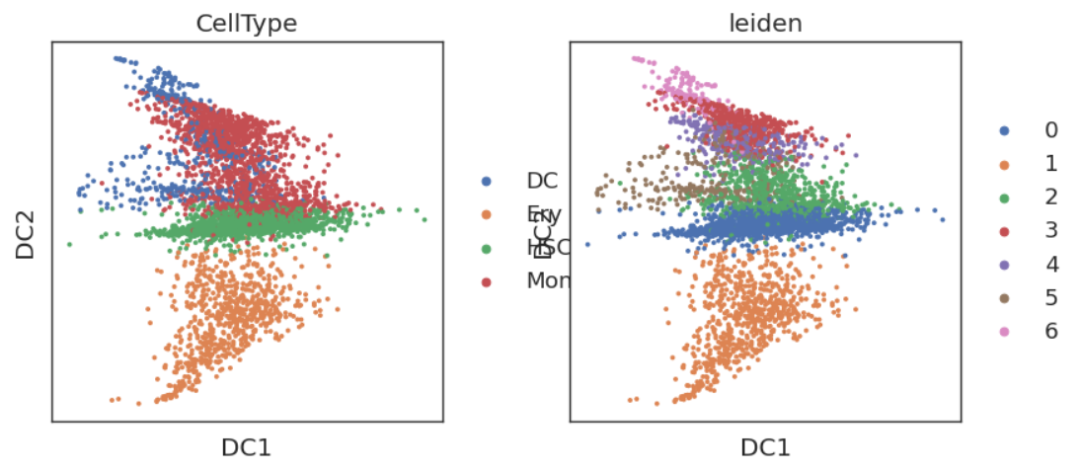
在这里我们可以清晰地看到,以HSC为中间点,向不同轨迹分化的方向。但我们也发现,DC1实际上没有编码分化的轨迹信息,因此我们可以尝试删除掉DC1:
adata.obsm['X_diffmap_'] = adata.obsm['X_diffmap'][:,1:]
sc.pl.embedding(adata,'diffmap_',color=['CellType','leiden'])
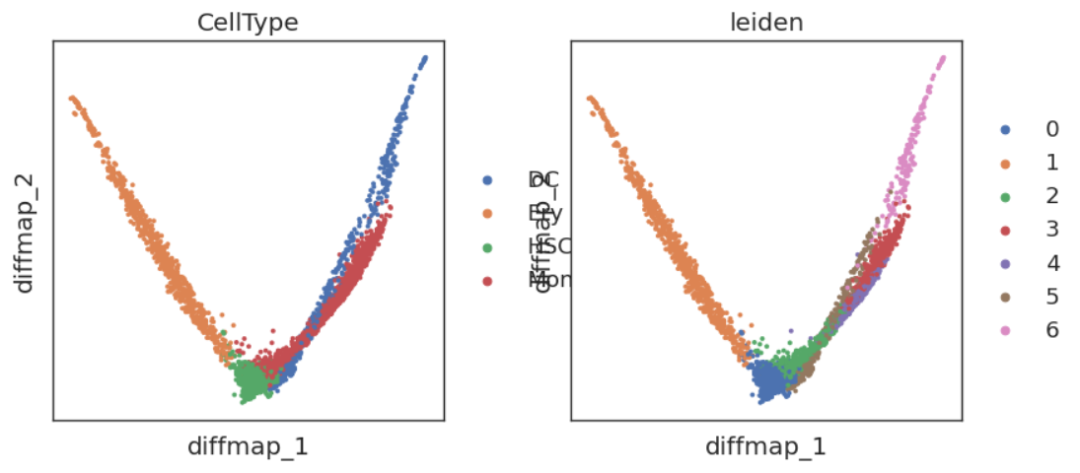
是不是有点Monocle内味了?但其实他们俩没啥关系。
我们再来看看FDG:
adata.obsm['X_diffmap'].shape
# (3821, 15)
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=15, use_rep='X_diffmap')
sc.tl.draw_graph(adata)
sc.pl.draw_graph(adata, color=['CellType'])

总体来说,需要根据实际情况选择用哪一种,DM的图形通常比较尖锐,而FDG更柔和。当细胞轨迹比较复杂多样时,DM通常会显得比较“紊乱”,这时候我们还可以这样来处理:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=10, use_rep='X_diffmap_')
sc.tl.umap(adata)
sc.pl.umap(adata,color='CellType')
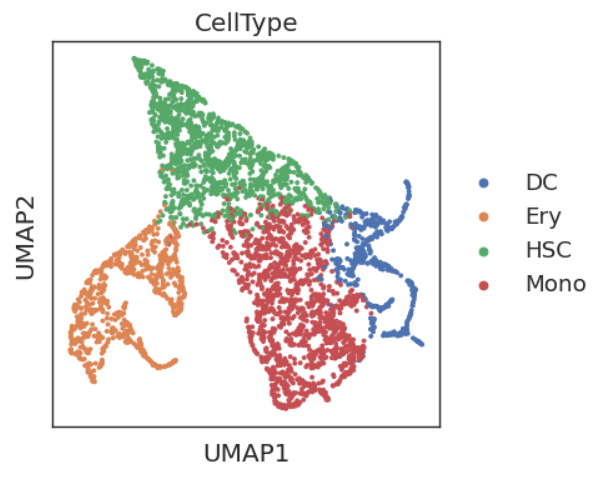
利用X_diffmap提供的embedding来替换PCA embedding,以获得更符合生物学轨迹的UMAP图。我们发现这个UMAP图更能把Mono和DC轨迹分开。此外,在上图中我们发现DC和Ery内部还有不同的分化方向。实际上基于先验知识的判断,它有一定的正确性,我们从下图的分化谱系中可以看到,DC细胞实际上有两个来源,其中一个是淋巴系分化出来,在UMAP表现为HSC直接分化得到,而另一个是由髓系分化,在UMAP图中表现为Monoctye分化而来。
。
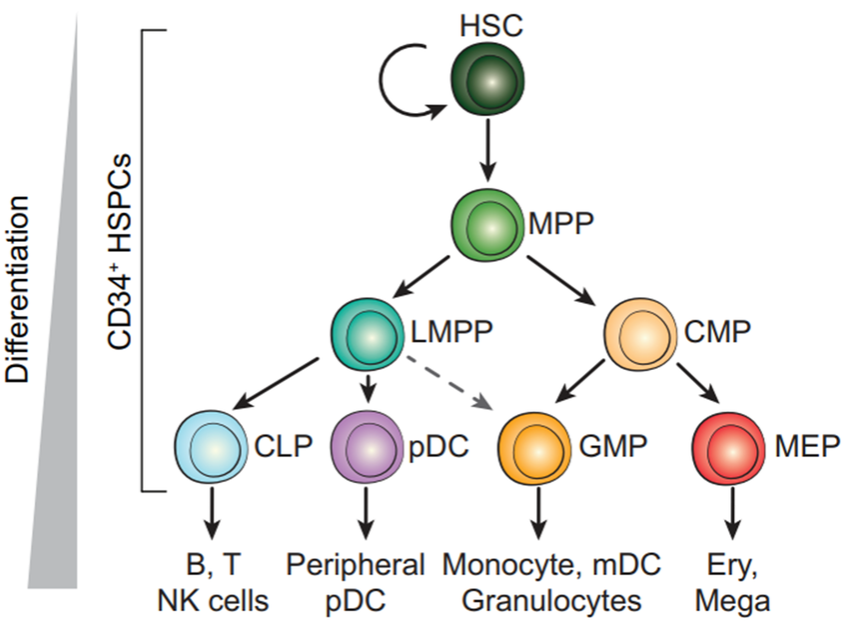
Palantir
Palantir可以认为提供root cell 和 terminal cells,我们回到Seurat中,利用CellSelector这个函数来精准选择细胞。
# anndata对象转换为h5这个通用格式
import diopy
diopy.output.write_h5(adata, 'diffmap.h5')
# Run in R
library(dior)
scobj = dior::read_h5('diffmap.h5')
scobj
Idents(scobj) = 'CellType'
p = DimPlot(scobj, reduction = 'diffmap_')
p
CellSelector(plot = p)
# 选择其中一个细胞
root.cell = 'Run4_130184147004718'
# 确认
DimPlot(scobj, cells.highlight = root.cell, reduction = 'diffmap_')
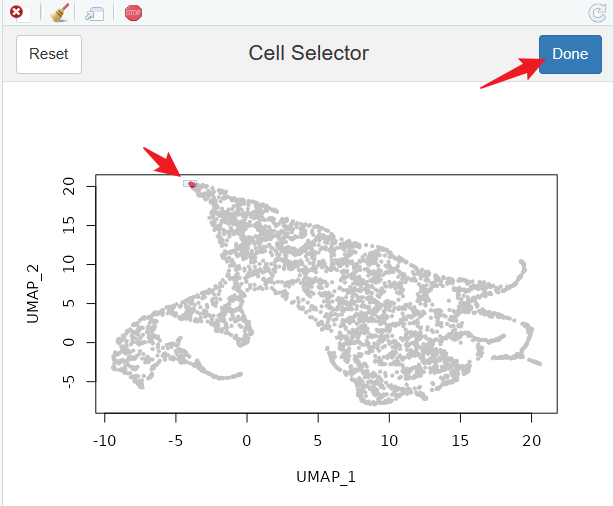
接下来回到Python中
## Palantir需要基于PCA
pca_projections = pd.DataFrame(adata.obsm['X_pca'], index=adata.obs_names)
## Palantir内置了diffusion map函数
dm_res = palantir.utils.run_diffusion_maps(pca_projections, n_components=10)
# Determing nearest neighbor graph...
# computing neighbors
# finished: added to `.uns['neighbors']`
# `.obsp['distances']`, distances for each pair of neighbors
# `.obsp['connectivities']`, weighted adjacency matrix (0:00:01)
dm_res['EigenVectors'].shape # embedding
# (3821, 10)
# Palantir内置函数帮助选择多少维DCs纳入分析
ms_data = palantir.utils.determine_multiscale_space(dm_res)
# 如果不选择:run me
# ms_data = dm_res['EigenVectors']
ms_data.shape
# (3821, 3)
运行:
# root cell
root_cell = 'Run4_130184147004718'
pr_res = palantir.core.run_palantir(ms_data,
early_cell=root_cell,
use_early_cell_as_start=True,
num_waypoints=500, knn=30)
获取信息和画图:
adata.obs['pseudotime'] = pr_res.pseudotime
adata.obs["DP"] = pr_res.entropy
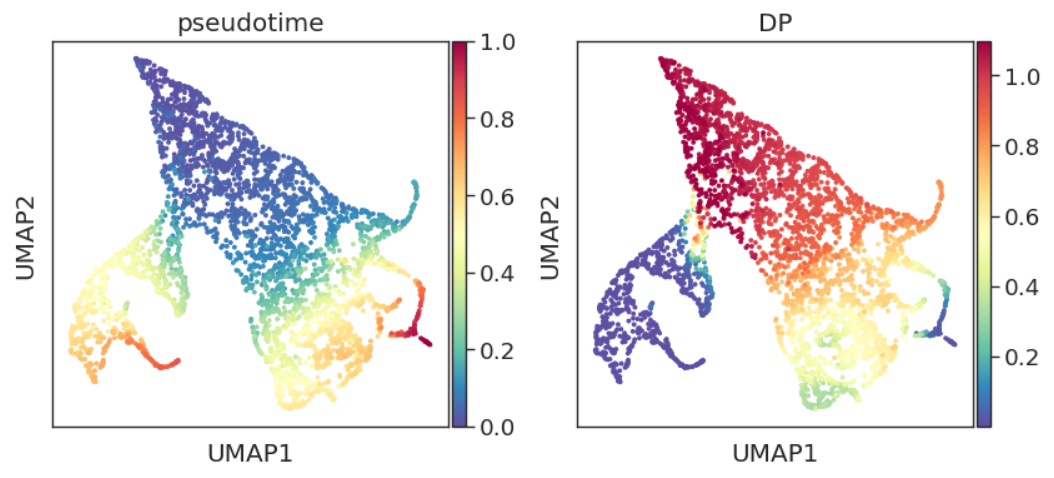
sc.pl.embedding(adata, basis="umap", color=['pseudotime', 'DP'])
DP即differential potential,通常与pseudotime呈反比。概念上与开头提到的“每个细胞状态分配了分化到不同终端状态的概率(Cell fate probabilities)”类似。结合先验知识,我们发现DP的结果与其生物学意义还是非常符合的。
此外,Palantir会提供自动计算的终点细胞名称。
pr_res.branch_probs.columns
# Index(['Run4_122436474493213', 'Run4_226265800857309', 'Run5_160996525231478'], dtype='object')
我们来看看这三个终点位置在哪里:
DimPlot(scobj,
reduction = 'umap',
cells.highlight = c('Run4_122436474493213', 'Run4_226265800857309', 'Run5_160996525231478'))
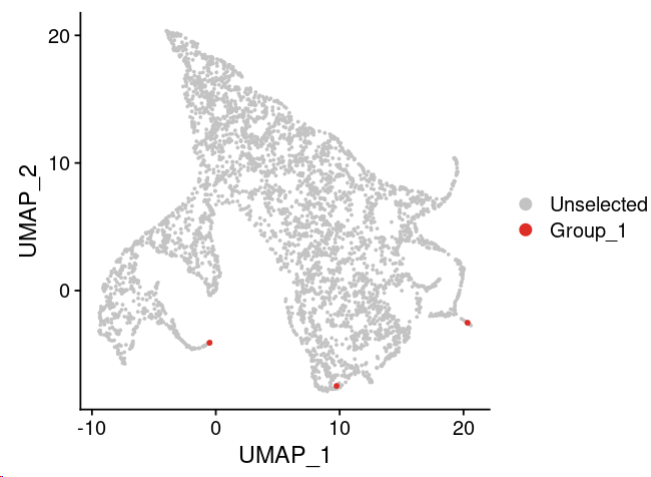
还是非常精确的!有时候如果觉得不满意,或者你还有其他希望人为指定终点的话,可以使用下面的例子。
我们再次从Seurat中选取终点细胞。假设我们希望针对DC潜在的另一个谱系分化进行研究。
DC_2 = 'Run4_236768101062563'

terminal_cells = {
"Run4_122436474493213": "Mono",
"Run4_226265800857309": "Ery",
"Run5_160996525231478": "DC1",
"Run4_236768101062563": "DC2",
}
pr_res = palantir.core.run_palantir(ms_data,
early_cell=root_cell,
use_early_cell_as_start=True,
num_waypoints=500,
terminal_states=terminal_cells)
pr_res.branch_probs.columns = ['Mono','Ery','DC1','DC2']
pr_res.branch_probs.head()
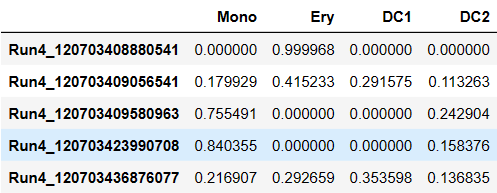
adata.obs['pseudotime'] = pr_res.pseudotime
adata.obs["DP"] = pr_res.entropy
sc.pl.embedding(adata, basis="umap", color=['pseudotime', 'DP'])
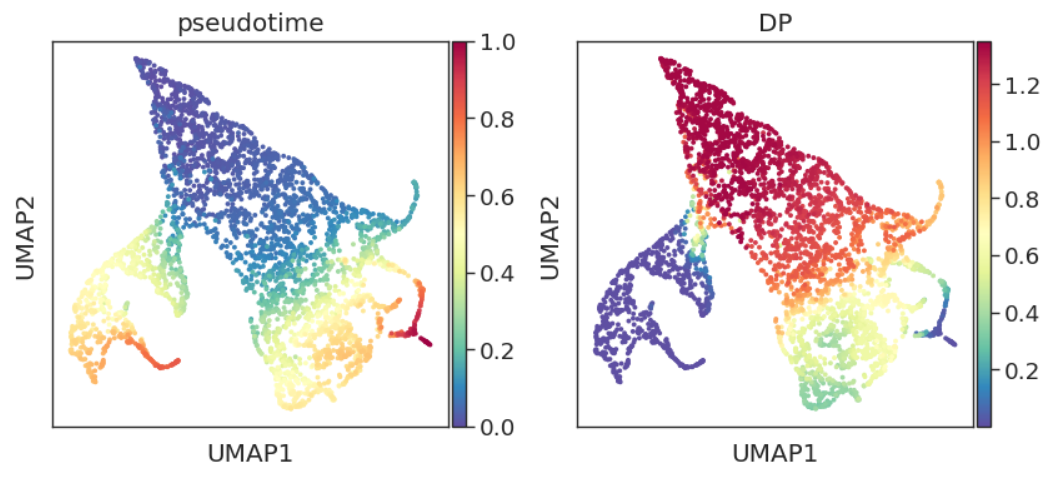
可以看到,结果还是与之前类似,尽管我们指定了DC2终点,但Palantir没有按照我们指定的细胞强行拟合。我们看看branch probability结果:

Palantir计算出来的几个谱系的界限和路径都非常清晰。
我们保存一下Palantir其他重要结果:
with open("tmp/palantir_waypoint.txt", "w") as fo:
[fo.write(cellID+"\n") for cellID in pr_res.waypoints.tolist()]
adata.obs.to_csv("tmp/palantir_results.csv")
导出为h5通用格式
diopy.output.write_h5(adata, 'palantir.h5')
小结
Palantir给我们提供了准确有效的拟时序信息和细胞干性评估,这对于后续分析至关重要,尤其是涉及细胞亚群表型转换的研究分析。如果你希望画出类似Slingshot的基因变化图,也可以参照之前我们分享过的笔记,利用Seurat和ggplot2画图体系来解决。事实上轨迹分析给我们添加了“时间”的维度,就会让很多事情变得有趣起来,就像3G网络变4G网络一样,以后我们再找时间继续分享轨迹分析的妙用吧!