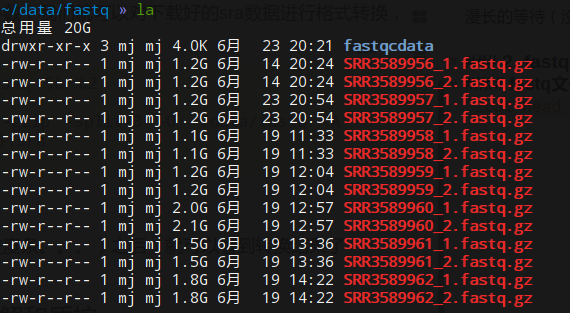
1.sra文件转换为fastq格式
为了进行测序数据质量检查我们需要将下载好的sra数据转换为fastq格式:使用Sratoolkits中的fastq-dump命令进行格式转换

Sratoolkits的官方文档中有fastq-dump命令的介绍(https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc&f=fastq-dump),fastq-dump的用法是fastq-dump [各种参数] <输入文件的路径>

- 主要会用到的参数有:
-O 指定输出路径
–gzip 指定输出格式为gzip压缩格式(fastqc软件可以直接识别gzip压缩的文件)
–bzip2 指定输出格式为bzip2压缩格式 (bzip格式较之gzip格式压缩效率更高,但是速度较慢)
多个文件参数
–split-3 如果是双端测序数据,则输出两个文件,如果不是则只输出一个文件。
fastq-dump -gzip -split-3 -O path -A file了解了命令用法和参数,我们就可以对下载好的sra数据进行格式转换,
for i in `seq 56 62`
do
fastq-dump -gzip -split-3 -O ~/data/fastq -A SRR35899${i}.sra
done漫长的等待(没有进度条。。。)之后,就可以看到转换结果了
2. fastq文件了解和质控
2.1 fastq文件格式
FASTQ文件每个序列分为四行,用以下命令打开一个fastq.gz的前四行
zcat path |head -n 4
@SRR3589956.1 D5VG2KN1:224:C4VAYACXX:5:1101:1159:2173 length=51
GGCGAGTGTAGGGCTGGCGCTGCCGGACGCGGTGCTAGTCGCCGGATGAAG
+SRR3589956.1 D5VG2KN1:224:C4VAYACXX:5:1101:1159:2173 length=51
B<BFBFBF0BFFFBFFBBFFIF<FFI<7<<BF<FFFFFFBB<BBBBBBBBB其中第一行以@开头,包含设备名称、run id等序列信息与相关的描述信息
第二行是碱基序列
第三行以+开头,可能包含序列信息与相关的描述信息或者没有描述
第四行是质量信息,与第二行的每一个碱基配对,表示其测序质量
2.2 使用fastqc进行质控
转换好的fastq文件不需要解压,可以直接使用fastqc进行质控:
* fastqc的常用参数有:
-o: 输出路径-
-extract:输出文件是否需要自动解压 默认是–noextract-
-t:线程数,和电脑配置有关,每个线程需要250MB的内存
-c:测序中可能会有污染, 比如说混入其他物种
-a:接头
-q:安静模式
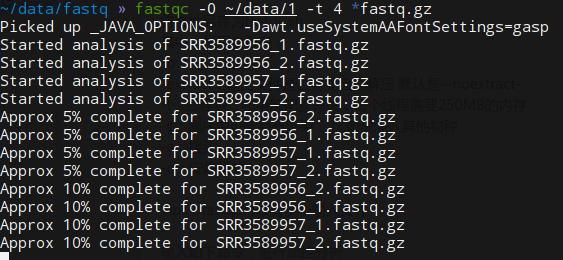
了解参数之后,可以用fastqc -O path -t n *.fastq.gz进行分析
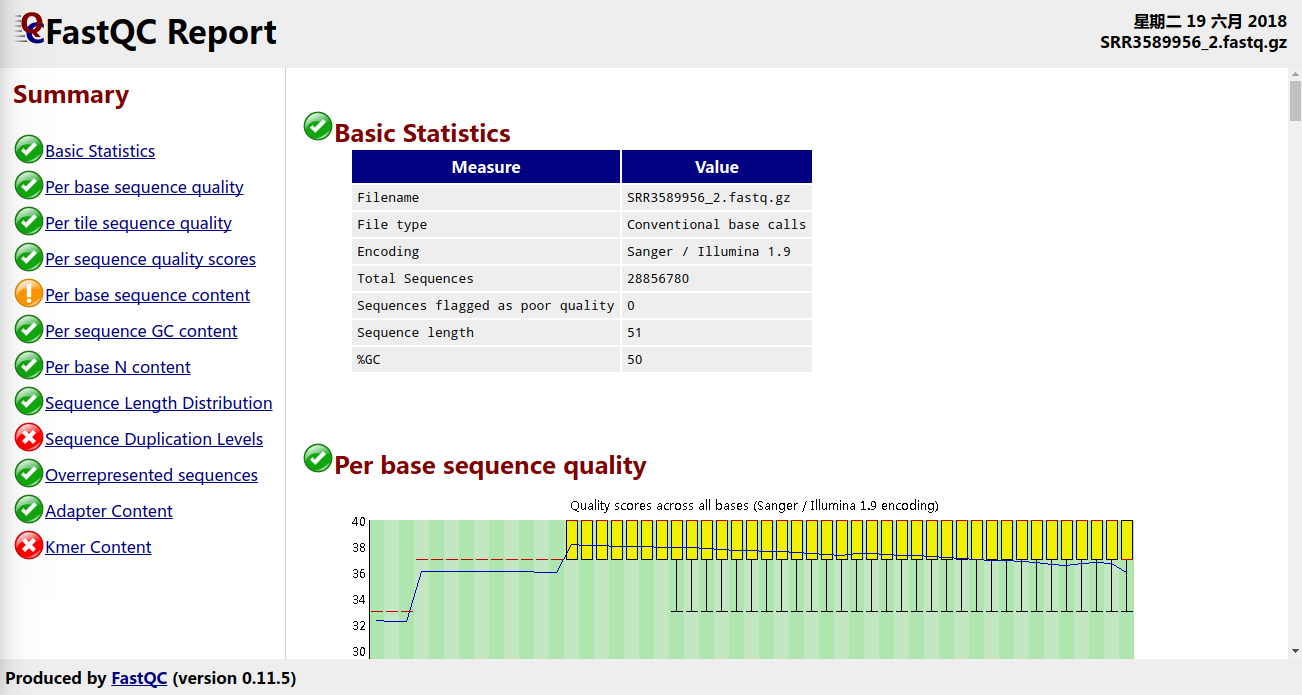
分析结束之后,得到一个fastqc.html文件和fastqc.zip文件,使用浏览器打开html文件即可直观看到质控结果
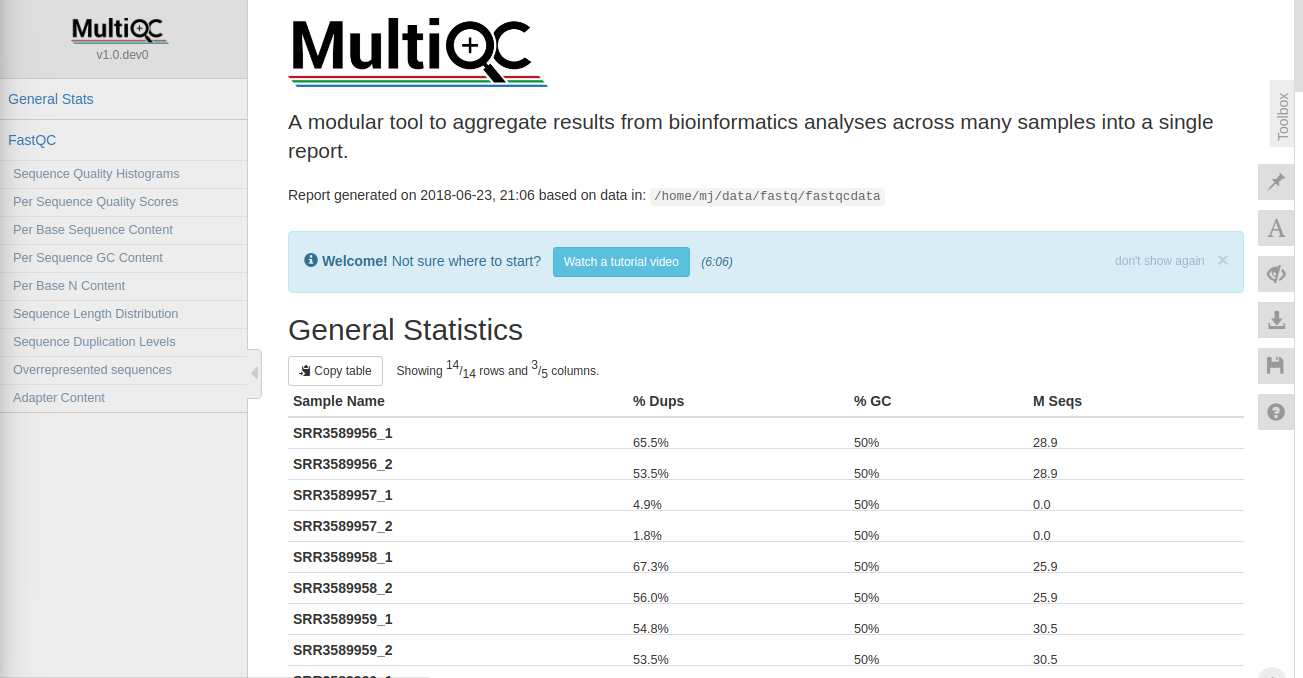
2.3 使用multiqc进行质控
multiqc可以对多个测序结果的qc结果进行整合,整理成一个报告。支持fastqc、trimmomatic、bowtie、STAR等多种软件。在安装了conda的情况下conda install -c bioconda multiqc即可安装
安装完毕后在需要分析的测序文件所在的文件夹multiqc .即可进行分析
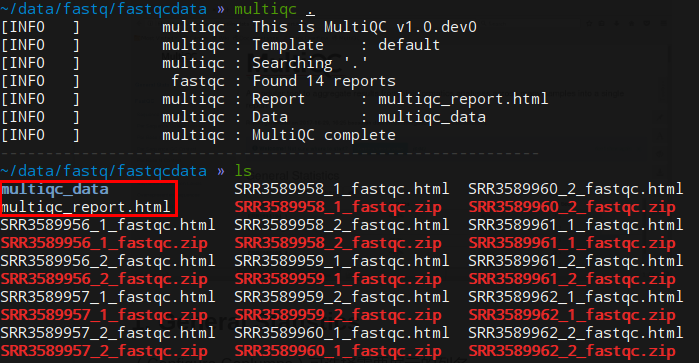
如需忽略某些文件,使用“–ignore”参数即可multiqc --ignore flie .