计算两个小分子之间的RMSD,可以用来判断两个构象的接近程度。
RDKit:计算不同分子或构象之间的RMSD点击打开链接
第一步:安装Anaconda
Win或者Linux系统下Anaconda安装,不赘述,网上很多教程。
第二步:安装RDKit
通过conda安装RDKit
conda install -c rdkit rdkit
第三步:使用方法
python isoRMSD.py mol1.pdb mol2.pdb rmsd.txt************************************************isoRMSD.py**********************************************
#!/usr/bin/python ''' calculates RMSD differences between 2 conformation with different atom names. @author: JC <[email protected]> ''' import os import sys import math # rdkit imports from rdkit import Chem from rdkit.Chem import AllChem from rdkit.Chem.AllChem import AlignMol def GetBestRMSD(probe,ref,refConfId=-1,probeConfId=-1,maps=None): """ Returns the optimal RMS for aligning two molecules, taking symmetry into account. As a side-effect, the probe molecule is left in the aligned state. Arguments: - ref: the reference molecule - probe: the molecule to be aligned to the reference - refConfId: (optional) reference conformation to use - probeConfId: (optional) probe conformation to use - maps: (optional) a list of lists of (probeAtomId,refAtomId) tuples with the atom-atom mappings of the two molecules. If not provided, these will be generated using a substructure search. Note: This function will attempt to align all permutations of matching atom orders in both molecules, for some molecules it will lead to 'combinatorial explosion' especially if hydrogens are present. Use 'rdkit.Chem.AllChem.AlignMol' to align molecules without changing the atom order. """ # When mapping the coordinate of probe will changed!!! ref.pos = orginXYZ(ref) probe.pos = orginXYZ(probe) if not maps: matches = ref.GetSubstructMatches(probe,uniquify=False) if not matches: raise ValueError('mol %s does not match mol %s'%(ref.GetProp('_Name'), probe.GetProp('_Name'))) if len(matches) > 1e6: warnings.warn("{} matches detected for molecule {}, this may lead to a performance slowdown.".format(len(matches), probe.GetProp('_Name'))) maps = [list(enumerate(match)) for match in matches] bestRMS=1000.0 bestRMSD = 1000.0 for amap in maps: rms=AlignMol(probe,ref,probeConfId,refConfId,atomMap=amap) rmsd = RMSD(probe,ref,amap) if rmsd<bestRMSD: bestRMSD = rmsd if rms<bestRMS: bestRMS=rms bestMap = amap # finally repeate the best alignment : if bestMap != amap: AlignMol(probe,ref,probeConfId,refConfId,atomMap=bestMap) return bestRMS, bestRMSD # Map is probe -> ref # [(1:3),(2:5),...,(10,1)] def RMSD(probe,ref,amap): rmsd = 0.0 # print(amap) atomNum = ref.GetNumAtoms() + 0.0 for (pi,ri) in amap: posp = probe.pos[pi] posf = ref.pos[ri] rmsd += dist_2(posp,posf) rmsd = math.sqrt(rmsd/atomNum) return rmsd def dist_2(atoma_xyz, atomb_xyz): dis2 = 0.0 for i, j in zip(atoma_xyz,atomb_xyz): dis2 += (i -j)**2 return dis2 def orginXYZ(mol): mol_pos={} for i in range(0,mol.GetNumAtoms()): pos = mol.GetConformer().GetAtomPosition(i) mol_pos[i] = pos return mol_pos if __name__ == "__main__": usage=""" isoRMSD.py will output two RMSD, one is fitted, another is no fit. Not fit RMSD mean no change in molecules coordinates. Usage:python isoRMSD.py mol1.pdb mol2.pdb rmsd.txt """ if len(sys.argv) < 4: print(usage) sys.exit() ref = Chem.MolFromPDBFile(sys.argv[1]) probe = Chem.MolFromPDBFile(sys.argv[2]) # here, rms is Fitted, rmsd is NOT Fit!!! rms,rmsd = GetBestRMSD(probe,ref) print("\nBest_RMSD: %.3f\nBest_Not_Fit_RMSD: %.3f\n"%(rms,rmsd)) out = open(sys.argv[3],"w") out.write("Best_RMSD: %.3f\nBest_Not_Fit_RMSD: %.3f\n"%(rms,rmsd)) out.close()
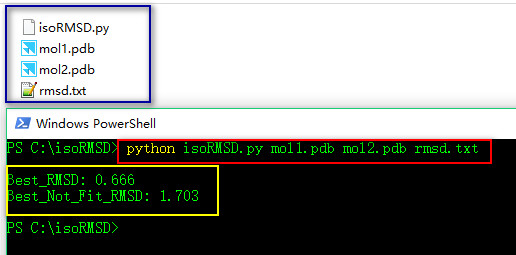
**********************************************************************************************************
参考链接:
https://github.com/0ut0fcontrol/isoRMSD